


Verfasst von: Desitin Redaktionsteam
Wenn jemand einen Schrei ausstößt, bewusstlos zu Boden geht und sich dann der ganze Körper verkrampft und zuckt, denken die meisten Menschen sofort an eine Epilepsie. Jedoch entspricht dieses weit verbreitete Bild nur einer ganz bestimmten Anfallsform, nämlich dem Grand-mal-Anfall. Und diese Form ist vergleichsweise selten. Weitaus weniger verbreitet ist die Tatsache, dass es verschiedenste Formen und Symptome epileptischer Anfälle gibt und manche sich sogar nur durch ganz subtile Anzeichen, zum Beispiel durch eine kurze geistige Abwesenheit der Betroffenen, Übelkeit oder Schwindel, bemerkbar machen.
Die typische Absence-Epilepsie etwa äußert sich mitunter dadurch, dass Betroffene für wenige Sekunden ihre aktuelle Tätigkeit unterbrechen, starr in die Luft gucken und dann wieder ganz normal mit ihrer vorherigen Aktivität fortfahren. Meistens können Betroffene selbst sich gar nicht an die kurze Absence erinnern. Diese Form der Epilepsie ist übrigens typisch für Kinder, weshalb sie oft als Träumerei oder Unkonzentriertheit fehlinterpretiert wird („Hans-guck-in-die-Luft“). Auch Schweißausbrüche, Halluzinationen, ein aufsteigendes Unwohlsein in der Magengegend und Herzrasen können Symptome epileptischer Anfälle sein.
Bei einer Epilepsie kommt es durch unterschiedlichste Ursachen und Auslöser zu einer übermäßigen elektrischen Entladung von Nervenzellen im Gehirn. So können zum Beispiel Stoffwechselstörungen, genetische Faktoren, Kopfverletzungen, gutartige und bösartige Tumore, Hirnhautentzündungen oder Schlaganfälle entsprechende Veränderungen im Gehirn verursachen, welche solche übermäßigen Entladungen der Neuronen begünstigen. Dann kommt es zu Symptome wie Muskelkrämpfen, Stürzen und Bewusstlosigkeit, aber auch zu durchaus subtileren Anfallsformen. Oft ist die genaue Ursache jedoch unbekannt. Das „Gewitter im Gehirn“ betrifft entweder Teilbereiche des Gehirns (fokale Epilepsie) oder das gesamte Gehirn (generalisierte Epilepsie). Bestimmte Abläufe, Häufigkeiten und Symptome werden zu sogenannten Epilepsie-Syndromen zusammengefasst, etwa der Juvenilen Absence-Epilepsie, dem Dravet-Syndrom oder der Rolando-Epilepsie. Zudem ist nicht jeder einmalige Krampfanfall gleichbedeutend mit einer Epilepsie. Im Kindesalter ist die Prognose außerdem sehr gut, sodass nicht jeder einmalige Anfall sofort zu einer dauerhaften Therapie führen muss. Die Prognose bezüglich Anfallsfreiheit variiert jedoch stark.
Der Begriff „Epilepsie“ beschreibt demnach das Auftreten oder das Risiko für das Auftreten mehrerer epileptischer Anfälle in bestimmten zeitlichen Abständen, während ein einmaliger epileptischer Anfall nicht zwangsläufig bedeutet, dass auch eine Epilepsie vorliegt, die mit Anfallssuppressiva behandelt werden muss.
Fokale Anfälle werden auch als partielle oder lokalisationsbezogene epileptische Anfälle bezeichnet. Diese Anfälle gehen immer von einem bestimmten Bereich des Gehirns aus und betreffen in der Regel nur eine Gehirnhälfte. Man unterscheidet fokale Anfälle mit Bewusstseinseinschränkung (früher auch komplex-fokal genannt) und fokale Anfälle ohne Bewusstseinseinschränkung (früher einfach fokale Anfälle). Im ersten Fall nimmt der Patient oder die Patientin den epileptischen Anfall nicht bewusst wahr und kann sich später an nichts erinnern. Bei Erwachsenen ist dies die am häufigsten beobachtete Anfallsform. Fokale epileptische Anfälle, vor allem solche mit Bewusstseinsstörung, können in einen sogenannten sekundär generalisierten Anfall (auch bilateral tonisch-klonischer Anfall) übergehen, der dann beide Gehirnhälften betrifft.
Die Symptome fokaler Anfälle richten sich nach dem Ursprungsort im Gehirn. Eine häufige Anfallsform fokalen Ursprungs sind vegetative fokale Anfälle.
Folgende Symptome sind typisch für fokale Epilepsien:
Auch plötzliche Angst, Wut oder Halluzinationen werden in der Literatur beschrieben. Die Sinneswahrnehmung kann durch einen fokalen Anfall gestört werden. So kann Sehen, Hören, Schmecken, Riechen oder Tasten durch den Anfall so beeinträchtigt sein, dass Betroffene Blitze sehen, Geräusche oder Stimmen hören, einen komischen Geschmack im Mund haben, etwas Merkwürdiges riechen oder Temperatur-Missempfindungen, Kribbeln oder Lähmungserscheinungen spüren. Fokale Anfälle mit Bewusstseinsverlust sind häufig durch sogenannte Automatismen geprägt. Patienten wiederholen im Anfall bestimmte Handlungsmuster, wie z. B. Räuspern, Schmatzen, bestimmte Sätze oder Fragen.
Bei generalisierten Anfällen lässt sich keine bestimmte Hirnregion zuordnen, in der der epileptische Anfall entsteht. Während eines Anfalls kann die Ausbreitung unterschiedlich verlaufen und das gesamte Hirnareal betreffen. Generalisierte epileptische Anfälle treten in verschiedenen Unterformen auf:
Bei Absencen kommt es zu einer plötzlichen Bewusstseinsstörung, sodass der Patient bzw. die Patientin seine oder ihre momentane Tätigkeit für die Dauer des Anfalls unterbricht. Die Betroffenen starren bei dieser Form eines epileptischen Anfalls oft ins Leere. Diese Anfälle können mehrere Sekunden dauern und sich stark gehäuft über den Tag wiederholen. Betroffene können sich an den Anfall nicht erinnern und fahren mit ihrer Tätigkeit nach dem Anfall wieder fort. Obwohl diese Anzeichen typisch für Absencen sind, werden sie von Laien vielfach nicht als Symptome einer Epilepsie erkannt. Absencen sind eine häufige Epilepsie-Form des Kindesalters und werden zunächst meist als Unkonzentriertheit oder Träumerei missinterpretiert. Es kann zu wenigen Anfällen innerhalb eines Jahres bis hin zu mehrenden hundert am Tag kommen. Wie oft epileptische Anfälle dieser Art auftreten, hängt vom individuellen Krankheitsbild ab.
Ein myoklonischer Anfall verursacht keine Bewusstseinsstörungen, sondern äußert sich mit Muskelzuckungen. Diese betreffen häufig Kopf und Arme, es können aber auch die Beine beteiligt sein.
Bei einem klonischen Anfall treten länger anhaltende rhythmische Zuckungen auf.
Tonische Anfälle sind gekennzeichnet durch andauernde Muskelanspannungen, die länger anhalten können.
Der tonisch-klonische Anfall oder auch Grand-mal-Anfall ist die Anfallsform, die am häufigsten mit der Krankheit Epilepsie in Verbindung gebracht wird. Die Symptome dieses Anfalls äußern sich meist in einem initialen Schrei des Betroffenen, gefolgt von einer Anspannung der Körpermuskulatur, die dann in Zuckungen des Körpers über geht (siehe oben). Ferner kommt es zu einem Bewusstseinsverlust, sodass sich der Patient bzw. die Patientin im Nachhinein nicht mehr an den Anfall erinnern kann. Auch die Blaufärbung der Lippen ist typisch. Sie entsteht durch die Verkrampfung der Atemmuskulatur während des Anfalls, sodass der oder die Betroffene keine Luft bekommt. Der Atemstillstand kann bis zu 30 Sekunden andauern, führt aber nicht zum Ersticken. Im Anschluss an einen Grand-mal-Anfall sind die meisten Patienten sehr erschöpft und müssen sich erholen.
Verliert man die Muskelkraft, spricht man von einem atonischen Anfall. Diese epileptischen Anfälle resultieren häufig in Stürzen.

„Die Epilepsie gilt als die am besten zu behandelnde Erkrankung der Neurologie.“
— Prof. Dr. Christian Elger, in der Folge 1 unseres Epilepsie 360° Podcasts für Patientinnen und Patienten
Die Symptome einer Epilepsie treten meist ganz plötzlich und unvermittelt auf, weshalb es entscheidend ist, dass Angehörige genau wissen, wie man schnell und präzise Erste Hilfe während eines Anfalls leistet. Das kann Angehörigen und Betroffenen große Angst machen. Deshalb wird häufig nach Vorboten gesucht, die einen epileptischen Anfall im Voraus ankündigen, um nicht davon überrascht zu werden, zum Beispiel beim Schwimmen oder Baden, wo selbst ein vergleichsweise kleiner epileptischer Anfall gefährlich werden kann. Und gelegentlich kündigen sie sich tatsächlich durch Anzeichen wie Kopfschmerzen, Schwindel, Stimmungsschwankungen und erhöhte Reizbarkeit bzw. Aggression der Patientinnen und Patienten an. Teilweise sogar schon Tage im Voraus. Unter diesen Umständen sollten Patientinnen und Patienten dann potenziell gefährliche Situationen wie das Autofahren, Wandern oder Schwimmen zeitweise meiden. Früher wurden diese Vorboten, ähnlich wie bei der Migräne, als Aura bezeichnet. Heutzutage weiß man, dass eine Aura selbst eine eigene Form epileptischer Anfälle ist. Die Vorboten, die aber keinesfalls allgemeingültig sind und nicht bei allen Patientinnen und Patienten auftreten, werden mittlerweile als Prodrom bezeichnet.
Ebenso wenig gibt es allgemeine Auslöser oder Trigger für epileptische Anfälle, deren Vermeidung dazu führen würde, dass es fortan zu keinen Epilepsie-Symptomen mehr kommt. Lange nicht alle Epileptiker*innen reagieren etwa mit Anfällen auf blitzendes oder flackerndes Licht. Doch auch hier gibt es große Unterschiede zwischen den einzelnen Formen einer Epilepsie.
Das alles muss Ihnen jedoch keine Angst machen. Denn meistens sind epileptische Anfälle zwar erschreckend, aber nicht gefährlich. Zumindest dann nicht, wenn sie gut kontrolliert und behandelt werden. Weder drohen durch akute Anfälle Hirnschäden, noch führt ein Anfall selbst zum Tod. Plötzliche und unerwartete Todesfälle durch einen epileptischen Anfall (SUDEP) sind sehr selten. Die weitaus größere Gefahr sind plötzliche Anfälle während des Autofahrens oder Stürze aus großer Höhe. Aufklärung und Vorbeugung, sowie eine konsequente Einnahme der Medikamente, sind hier mit Abstand der beste Weg, dieses Risiko zu reduzieren.
Die Epilepsie gilt als eine der am besten zu behandelnden neurologischen Erkrankungen der Welt und bis zu zwei Drittel der Patientinnen und Patienten werden durch die medikamentöse Therapie mit Antikonvulsiva anfallsfrei. Da Epilepsie jedoch nicht heilbar ist, gilt die Anfallskontrolle als wichtigstes Ziel. Diese ist oft nur durch eine lebenslange Einnahme der Anfallssuppressiva möglich, welche dann aber oft ein uneingeschränktes und selbstständiges Leben bis ins hohe Alter ermöglicht.

Informationen für Ärztinnen und Ärzte
Fachinformationen, aktuelle Studiendaten
und vieles mehr zum Thema Epilepsie
Die übermäßige Aktivität der Neuronen kann zu diversen Störungen führen und sich mitunter auch als typischer motorischer Krampfanfall äußern. Das muss aber nicht so sein. Manche Anfälle werden sowohl von Patientinnen und Patienten als auch Angehörigen kaum wahrgenommen, während andere zu Bewusstlosigkeit, Muskelkrämpfen, Stürzen und Zittern führen können. Ebenso können Störungen des Geruchssinns, Halluzinationen, Wutausbrüche, Migräne und Übelkeit auftreten. Oder Patientinnen und Patienten springen plötzlich auf und werfen Stühle um oder rennen unkontrolliert umher, woran sie sich später nicht erinnern können.
Es können also eine Vielzahl unterschiedlicher Symptome und Anfallsformen auf eine Epilepsie hinweisen, was die Diagnose der Erkrankung deutlich erschwert. Sowohl die Dauer, als auch Form und Ausprägung der motorischen und nicht-motorischen Symptome können erheblich variieren. Manche Anfälle dauern nur wenige Sekunden, andere mehrere Minuten. Mal äußern sie sich durch das allseits bekannte Erscheinungsbild der verkrampfenden und zuckenden Muskulatur, andere wiederum äußern sich durch nicht-motorische Symptome. Die häufigste Anfallsform bei Erwachsenen sind komplex-fokale Anfälle, die mit Bewusstlosigkeit einhergehen.
Aufgrund der starken Unterschiede kann es auch für Fachärztinnen und Fachärzte schwierig sein, eine Epilepsie zu diagnostizieren und von anderen Formen von Krampfanfällen zu unterscheiden, insbesondere von psychogenen Krampfanfällen, Fieberkrämpfen und akut symptomatischen Krampfanfällen.
Zur Einteilung der sich stark unterscheidenden epileptischen Anfälle und ihrer individuellen Symptome wird die Definition der Internationalen Liga gegen Epilepsie (International League Against Epilepsy – ILAE) herangezogen.
Nach Definition der ILAE wird eine Epilepsie zunächst anhand ihres fokalen oder generalisierten Beginns klassifiziert:
Des Weiteren gibt es epileptische Anfälle, die keiner der genannten Klassen zuzuordnen sind. Diese gruppiert die ILAE als Anfälle mit mutmaßlicher Ursache (idiopathische Epilepsie). Das Feld der Ursachenforschung in Bezug auf epileptische Anfälle ist hier noch weit und wirft immer neue Fragestellungen auf.
Auch Gefühls- und Verhaltensänderungen können Teil eines epileptischen Anfalls sein und werden als Krankheitszeichen der Epilepsie verstanden.
Zwar sind die Anfallsformen vielfältig, jedoch treten bei einzelnen Epilepsie-Patientinnen und Patienten in der Regel nur ein bis maximal drei verschiedene Formen epileptischer Anfälle auf.
Treten bestimmte Symptome, Abläufe und aktivierte Gehirnregionen regelmäßig zusammen in Erscheinung, kann man diese zu fest definierten Epilepsiesyndromen zusammenfassen. Die Syndrome unterscheiden sich anhand der Epidemiologie (Verbreitung), der Klinik (Ablauf/Symptome des Anfalls) und weiterer Befunde in der Diagnose. Es gibt sowohl fokale Epilepsiesyndrome (z. B. Rolando-Epilepsie) als auch generalisierte Epilepsiesyndrome (z. B. Juvenile Absence Epilepsie), die wir zur besseren Übersicht an späterer Stelle in Tabellen für Sie zusammenfassen.
Mögliche Symptome eines epileptischen Anfalls:
Die verbreitete Vorstellung eines epileptischen Anfalls ist dramatisch: Die Patientinnen und Patienten stürzen und bekommen Krämpfe am ganzen Körper. Allerdings tritt diese Form des Anfalls in der Praxis eher selten auf. Das in der Bevölkerung verbreitete Bild des epileptischen Anfalls entspricht dem sogenannten „Grand mal“-Anfall (oder auch großem Krampfanfall, generalisiert-tonisch-klonischer Anfall). Neben dieser Anfallsform gibt es jedoch viele weitere Formen von Krampfanfällen, angefangen bei der Aura (Sinnesstörungen unterschiedlichster Art) bis hin zu sogenannten Absencen (kurzem Verlust des Bewusstseins), die teilweise nur wenige Sekunden dauern und mit keinerlei Muskelverkrampfungen oder Zuckungen einhergehen.
Zum Download: PDF Epilepsie Anfallsformen
Die ILAE definiert einmalige Anfälle als sogenannte akut symptomatische Anfälle. Sie stehen nicht direkt mit Epilepsie in Verbindung, sondern ähneln den epileptischen Anfällen nur. Die Ursachen sind hier jedoch andere. Sie treten in direktem Zusammenhang mit anderen Erkrankungen oder in akuten Krankheitssituationen auf, z. B. als Folge einer Unterzuckerung, Kopfverletzung, Vergiftung oder einer Hirnschädigung, sowie auch nach einem Schlaganfall und sind einmalige Ereignisse. Dazu zählen zum Beispiel auch die im Kindesalter häufig auftretenden Fieberkrämpfe.
Allerdings können Ursachen wie Schlaganfälle oder Verletzungen auch zu dauerhaften strukturellen Veränderungen im Gehirn führen, die fortan wiederholt epileptische Anfälle auslösen. Dann spricht man von einer strukturellen Epilepsie, die früher auch als symptomatische Epilepsie bezeichnet wurde. Dazu zählen auch epileptische Anfälle, welche durch Tumore ausgelöst werden. Führen Infektionen, etwa Hirnhautentzündungen, zu entsprechenden chronischen Veränderungen im Gehirn, so spricht man von infektiösen Epilepsien. Sind Stoffwechselstörungen der Grund, handelt es sich um eine metabolische Epilepsie. Weitaus häufiger ist aber keine klare Ursache identifizierbar. Dann handelt es sich um genetische oder kryptogene Epilepsien. In jungen Jahren sind zudem häufig andere Auslöser identifizierbar als im höheren Lebensalter.
Bei Jugendlichen und jungen Erwachsenen mit Epilepsie spielen oft Unfälle oder Tumore eine Rolle, bei älteren Menschen hingegen Durchblutungsstörungen, Schlaganfälle oder Degenerationsprozesse.
Bei Säuglingen und Kleinkindern können Schwangerschaftskomplikationen oder eine Störung der Entwicklung des Gehirns ursächlich sein. Zudem zeigen neuere Untersuchungen, dass es auch genetische Risikofaktoren für die Entwicklung einer Epilepsie gibt. So kann die genetische Veranlagung in einzelnen Familien zwar erhöht sein, vererbbar ist Epilepsie aber nicht.

Mögliche Ursachen & Auslöser einer Epilepsie
Ursachen einer Epilepsie: Die Suche nach dem Auslöser. Eines haben alle Epilepsien gemeinsam: Sie haben ihren Ursprung im Gehirn. Als Auslöser kommen jedoch verschiedenste Ursachen infrage.
Ein wichtiger Unterschied zwischen Epilepsie und Krampfanfall: Es gibt keinen akuten Auslöser für epileptische Anfälle, sondern eine dauerhafte Ursache wie eine strukturelle Veränderung im Gehirn oder eine chronische Grunderkrankung, die immer wieder zu epileptischen Anfällen führt. Anders formuliert bedeutet das, dass ein Vermeiden der Auslöser akut symptomatischer Krampfanfälle, zum Beispiel durch mehr Schlaf, weniger Stress, weniger Alkohol, oder das Auskurieren einer Fiebererkrankung, das Risiko für einen erneuten akuten Anfall minimieren kann. Das Risiko für den nächsten epileptischen Anfall kann meistens jedoch nicht signifikant durch die Vermeidung individueller Trigger reduziert werden, da die eigentliche Ursache eine andere ist, etwa eine prozesshafte Grunderkrankung oder eine veränderte Struktur der Hirnrinde. Hier gelingt eine Anfallskontrolle, bis hin zur Anfallsfreiheit, nur durch eine dauerhafte Behandlung, in erster Linie durch die lebenslange Einnahme von Anfallssuppressiva.

Abgrenzung epileptischer Anfälle zu anderen Krampfanfällen
Andere Anfälle Krampfanfall, Fieberkrampf oder Epilepsie – Die Unterschiede Der Ausdruck „epileptischer Anfall“ ist eine Sammelbezeichnung für unterschiedlichste Krankheitsbilder; dabei ist nicht jeder einmalige Anfall […]
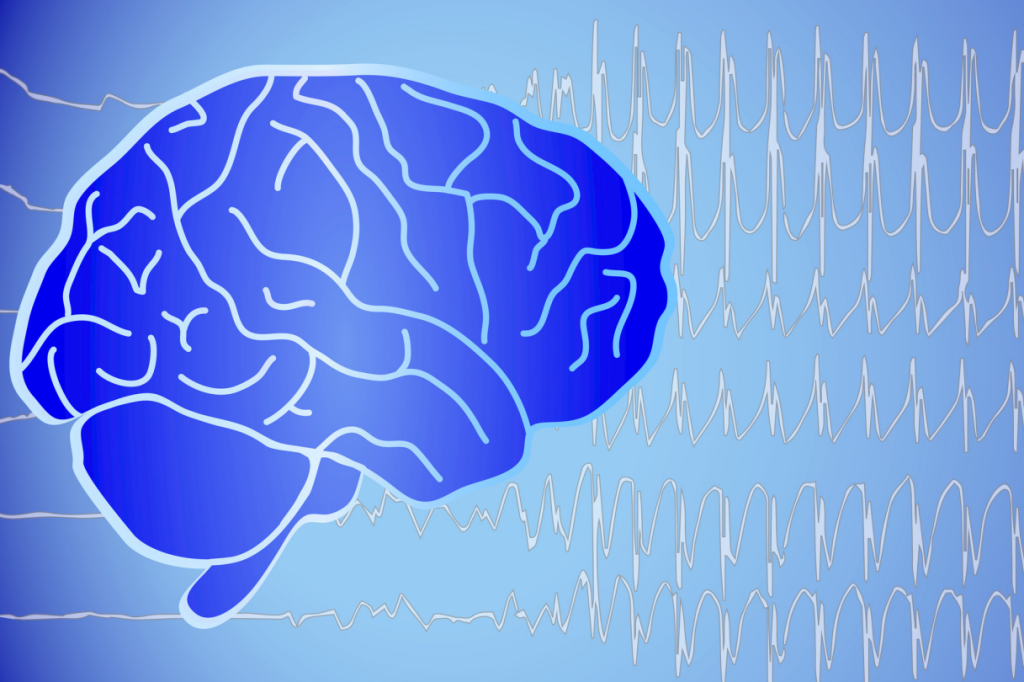
Die Diagnose einer Epilepsie kann komplex sein
Die Diagnose einer Epilepsie basiert auf vielen Säulen, von einer ausführlichen Anamnese, über jahrelange Erfahrung der Neurologen und Neurologinnen, bis hin zur Auswertung von EEG-Potenzialen und MRT.
Fast jedes zehnte Kind und 5 - 10% aller Menschen erleben mindestens einen epileptischen Anfall. In diesem Fall spricht man von sogenannten Gelegenheitsanfällen. Doch ein einmaliger Anfall ist noch nicht gleichbedeutend mit einer Epilepsie. Mindestens zwei epileptischen Anfällen ohne erkennbare Ursache erleben ca. 1% aller Menschen. Kinder und ältere Menschen sind häufiger betroffen; etwa 2/3 aller Epilepsien treten bis zum 20. Lebensjahr auf. Ab dem 60. Lebensjahr erhöht sich das Risiko, an Epilepsie zu erkranken, im Vergleich zum Erwachsenenalter dann erneut, bedingt durch z. B. Schlaganfälle, degenerative Alterungsprozesse, Hirntumore oder Demenzerkrankungen.
Ist die Diagnose erstmal gestellt, unterscheidet sich auch der Verlauf sehr stark von Patient*in zu Patient*in.
Man kann also nicht pauschal sagen, wie häufig Menschen mit Epilepsie Anfälle erleben, wie lange diese dauern und wie viel Zeit bis zum nächsten Anfall vergeht. Fest steht jedoch, dass epileptische Anfälle häufig gut behandelbar sind, wenn eine genaue Diagnose erfolgt ist. Und wie erschreckend das Anfallsgeschehen mitunter auch aussehen mag, gefährlich ist es für das Gehirn meistens nicht. Weder führen sie zum Absterben von Nervenzellen im Gehirn, noch beeinträchtigen sie langfristig die Gedächtnisleistung, wenngleich Patientinnen und Patienten während eines Anfalls das Bewusstsein verlieren und sich nicht an das Anfallsgeschehen erinnern können. Ebenso gibt es jedoch Anfälle, die von Betroffenen bei vollem Bewusstsein erlebt werden.
Auch die Lebenserwartung ist nicht grundsätzlich geringer als jene von Menschen ohne Epilepsie. Doch auch hier sind generelle Aussagen schwierig. Die Prognose, vor allem bezüglich der Anfallsfreiheit, variiert je nach Epilepsie-Form stark. Die Diagnose Epilepsie bedeutet aber nicht automatisch, dass Betroffene kein selbstbestimmtes und zufriedenes Leben führen können. Eine teilweise erhöhte Sterblichkeit im Vergleich zur gesunden Bevölkerung ist zudem meistens nicht direkt auf den epileptischen Anfall zurückzuführen, sondern viel mehr auf langfristige Konsequenzen für die psychische Verfassung (Depression, Suizidgefahr) und eine erhöhte Unfallgefahr. Zum Beispiel durch Stürze oder einen Krampfanfall beim Autofahren. Durch gezielte Maßnahmen zur Vorbeugung und Aufklärung rund um Epilepsie kann dieses Risiko erheblich gesenkt werden. Weiterhin werden einige Epilepsien durch ernsthafte Grunderkrankungen ausgelöst, zum Beispiel Tumore oder Stoffwechselstörungen. In diesen Fällen ist die erhöhte Sterblichkeit auf die eigentliche Grunderkrankung und nicht auf die epileptischen Anfälle zurückzuführen.
Geben Sie die Postleitzahl Ihres Wohnortes ein, um Epilepsie-Zentren in Ihrer Nähe zu finden.
Finden Sie Epilepsie-Zentren in Ihrer Nähe:
Der Ablauf eines epileptischen Anfalls ist sehr individuell und kann keinesfalls verallgemeinert werden. Jedoch gibt es bestimmte Epilepsiesyndrome und Anfallsformen, die ein gewisses Muster erkennen lassen. Die wichtigste Unterscheidung findet hier zwischen fokalen und generalisieren Epilepsien statt, die aber auch ineinander übergehen können. Und auch innerhalb dieser Klassifizierung können Anfälle völlig unterschiedlich beginnen und ablaufen.
Bei einigen Formen der Epilepsie gibt es jedoch Vorboten oder Anzeichen, die einen bevorstehenden epileptischen Anfall ankündigen können. Früher wurden diese auch als Aura bezeichnet. Heute weiß man, dass eine Aura, wie z. B. die epigastrische Aura, eigentlich kein Vorbote eines Anfalls ist, sondern eine eigene Anfallsform ist, ähnlich wie die Absencen. Diese besondere Art von fokalen Anfällen hält oft nur wenige Sekunden an. Zum Beispiel ein unangenehmes Gefühl in der Magengegend, welches bis zum Hals aufsteigt. Oder Patientinnen und Patienten haben Halluzinationen, beispielsweise nehmen sie Gerüche wahr, die gar nicht da sind. Auch ein gesteigertes Angstgefühl oder eine veränderte visuelle Wahrnehmung (Lichtblitze, bunte Punkte oder ein eingeschränktes Gesichtsfeld) können auftreten. Auren sind für Ärztinnen und Ärzte wichtige Anhaltspunkte dafür, die entsprechende Hirnregion einzugrenzen, in welcher die fokalen (herdförmigen) epileptischen Anfälle entstehen.
Vorboten oder Vorahnung eines epileptischen Anfalls bezeichnet man hingegen als Prodrom.
Diese Vorzeichen halten teilweise mehrere Tage an, bevor es zu einem epileptischen Anfall kommt. Sie können aber auch nur wenige Stunden oder Minuten im Voraus einen Anfall ankündigen, oder eben gar nicht auftreten. Einige Anfälle beginnen zum Beispiel auch mit einem leichten Kribbeln in den Fingern. Bei solchen Anzeichen kann es teilweise hilfreich sein, wenn man gezielt mit anderen Reizen gegensteuert. Zum Beispiel kann man dem Kribbeln in der Hand mit einer geballten Faust begegnen. So werden die entsprechenden Zellen im Gehirn bewusst anderweitig beschäftigt. Bei anderen Symptomen, zum Beispiel bei Gedankenkreisen oder der Wahrnehmung nicht vorhandener Gerüche kann es helfen, sich bewusst auf die Atmung zu konzentrieren oder aber Geruchsstimuli zu setzen, z. B. mit Aroma-Ölen. Gelegentlich ist es möglich, beginnende Anfälle so gezielt zu unterbrechen, was aber keinesfalls die Einnahme von Anfallssuppressiva ersetzt.
Sicher haben Sie sich schon Gedanken darüber gemacht, welche äußeren Umstände das Auftreten des
einzelnen Anfalls bei Ihrem Kind, sich selbst oder Angehörigen hervorrufen oder zumindest begünstigen könnten. Die Erfahrung hat gezeigt, dass in fast allen Fällen die epileptischen Anfälle unvorhersehbar – wie aus heiterem Himmel – auftreten.
Spricht man von Epilepsie, so haben viele Menschen das Bild eines Menschen im Kopf, der auf flackerndes Licht mit einem Bewusstseinsverlust reagiert, zu Boden fällt, die Augen verdreht und dann am ganzen Körper krampft oder zittert. Doch das ist nicht das typische Bild einer Epilepsie und lange nicht alle Epileptiker*innen reagieren auf bestimmte Trigger oder Auslöser. Viel mehr ist eines der entscheidenden Merkmale einer Epilepsie, dass Anfälle auch ohne erkennbaren äußeren Reiz bzw. Auslöser auftreten.
Dennoch gibt es auch für Epileptiker*innen Auslöser oder sog. Trigger, welche einen epileptischen Anfall begünstigen können und die man vermeiden sollte, um Betroffenen das Leben zu erleichtern und die Anfallskontrolle zu unterstützen. Diese Trigger sind jedoch sehr individuell und gelten nicht für alle Patientinnen und Patienten mit Epilepsie. Verkrampfungen und Zittern am ganzen Körper sind außerdem nicht die einzigen Symptome, die einen epileptischen Anfall beschreiben. Sie treten in diesem Ausmaß nur bei einem sog. „Grand-mal-Anfall“ auf. Im Alltag gibt es große Unterschiede zwischen epileptischen Anfällen und ebenso bei den möglichen Ursachen und Auslösern.
Wichtig zu verstehen ist, dass prinzipiell jedes Gehirn mit Anfällen reagieren kann, wenn es stark gereizt wird. Auslöser können zum Beispiel Übermüdung, extreme Witterungswechsel, fieberhafte Infekte, Sauerstoffmangel, Stress, Fieber, Entzündungen des Gehirns oder Kopfverletzungen sein. Auch Alkohol, Drogen und Vergiftungen können solche akut symptomatischen Anfälle auslösen, ebenso wie die Monatsblutung bei Frauen und Mädchen in seltenen Fällen Anfälle begünstigen kann. Hier spricht man jedoch zunächst nur von einem (akut-symptomatischen) Krampfanfall. Rund 10% der Menschen erleben mindestens einmal im Leben einen solchen Krampfanfall oder Fieberkrampf.
Jedoch sollten individuelle Auslöser eines epileptischen Anfalls auf keinen Fall mit den eigentlichen Ursachen der Erkrankung verwechselt werden, die zudem noch nicht vollständig erforscht sind.
Warum genau solche Trigger insbesondere die Häufigkeit von epileptischen Anfällen bei einigen Patientinnen und Patienten steigern können, ist nicht abschließend geklärt. Es steht aber fest, dass vor allem Stress den Hormonhaushalt empfindlich verändern und die Schlafqualität verschlechtern kann. Ausreichender Schlaf von mind. acht Stunden, sowie Tipps zum bewussten Stressmanagement können die Anfallskontrolle bei Epilepsie also mitunter entscheidend verbessern. Bezüglich der Schlafdauer ist es übrigens nicht wichtig, ob Sie nun von 22:00 Uhr bis 06:00 Uhr schlafen, oder von 03:00 Uhr morgens bis 11:00 Uhr. Entscheidender ist, dass der Schlaf-Wach-Rhythmus stabil ist und nicht von Tag zu Tag variiert. Mehr Tipps zum Alltag mit Epilepsie finden Sie ebenfalls aus unserer Website.
Vermeidbarer Auslöser Nummer eins ist das absichtliche Weglassen der verordneten Medikamente, ohne Absprache mit den behandelnden Ärztinnen und Ärzte. Angenehme oder unangenehme Aufregungen allein führen hingegen selten zu epileptischen Anfällen. Dennoch sollte man diese Zusammenhänge genau beobachten, um mögliche psychische und physische Trigger zu erkennen, um diese Situationen zukünftig zu meiden. Das kann epileptische Anfälle zwar nicht vollständig verhindern, aber die Anfallshäufigkeit reduzieren.
Bei manchen besonders veranlagten Kindern und Erwachsenen können auch bestimmte Sinnesreize aus unserem Alltag epileptische Anfälle auslösen, die auch als Trigger bezeichnet werden. Dazu gehören insbesondere optische Reize (Hell-Dunkel-Kontraste, Flackerlicht), sehr viel seltener aber auch überraschende Geräusche, Berührung oder Schreckmomente. Solche durch äußere Reize ausgelösten Anfälle nennt man Reflexanfälle. Von einer Epilepsie spricht man aber auch dann erst, wenn solche Anfälle auch ohne Reizauslösung auftreten (nicht-provozierte Anfälle).
Manche Menschen erholen sich nach epileptischen Anfällen in wenigen Sekunden und sind quasi sofort wieder fit. Es kann jedoch auch sein, dass die Betroffenen nach einem Anfall in einen tiefen Schlaf fallen, da der Körper sich erholen muss. Andere wiederum sind zwar schnell wieder ansprechbar, aber sehr verwirrt oder erschöpft. Teilweise können die Betroffenen sich an den Anfall erinnern, teilweise nicht.
Manche Menschen erleben nach einem Anfall auch längere Kopfschmerzen, Muskelschmerzen, Verwirrtheit oder depressive Stimmungen und benötigen Ruhe und Erholung, während andere sofort wieder in ihre vorherige Tätigkeit einsteigen können oder wollen. Es ist wichtig, das Bewusstsein und den Zustand der betroffenen Person zu überprüfen und bei ihr zu bleiben, bis sie wieder voll orientiert ist und sich sicher bewegen kann.
Es kann einige Zeit dauern, bis sich betroffene Personen wieder vollständig orientieren können. Daher sollte man ihnen Zeit geben und ihnen helfen, Erinnerungslücken zu schließen. Bei Bewusstlosigkeit ist es wichtig, die Person in die stabile Seitenlage zu bringen, um Aspiration zu vermeiden. Es ist hilfreich, die betroffene Person nach ihren Wünschen und Bedürfnissen zu fragen und darauf zu achten, welche Form der Unterstützung sie sich wünscht. Den Ablauf des Anfalls und die Aussagen der Patientinnen und Patienten sollten unbedingt dokumentiert werden, am besten in einem Anfallskalender. Für die Diagnose und Therapie der Epilepsie bzw. die Überprüfung des Behandlungserfolgs ist dies entscheidend.
Was Angehörige und Ersthelfer nach dem Anfall tun sollten:
Mögliche Folgen eines epileptischen Anfalls
Epileptische Anfälle führen normalerweise nicht zu dauerhaften Gehirnschäden oder geistigen Beeinträchtigungen. Allerdings kann der Status epilepticus dauerhafte Schäden verursachen. Ebenso kann es während eines Anfalls in sehr seltenen Fällen auch zu einem plötzlichen Tod kommen, was dann als SUDEP bezeichnet wird.
Die unmittelbaren Folgen eines epileptischen Anfalls variieren je nach Person und Anfallsstärke.
Folgen im Überblick:
Was Sie unbedingt über SUDEP wissen sollten
SUDEP – der plötzliche Tod durch Epilepsie und was Sie darüber wissen sollten Das Wichtigste im Überblick Was bedeutet SUDEP? SUDEP ist die Abkürzung für […]
Erste Hilfe bei epileptischen Anfällen
Erste Hilfe bei epileptischen Anfällen Ein epileptischer Anfall kann auf den ersten Blick angsteinflößend aussehen. Deshalb ist es wichtig zu verstehen, was genau Patientinnen und […]
Unser Desitin Magazin
Unsere Fachartikel für Ärztinnen und Ärzte zum Thema Epilepsie und weitere interessante Informationen

Generell gibt es verschiedenste Arten epileptischer Anfälle. Sie werden auf Basis ihrer konkreten Symptome und der Hirnregionen klassifiziert, in denen das Anfallsgeschehen beginnt, was bei einem EEG festgestellt werden kann. Es gibt vor allem zwei Arten von Anfällen:
Epileptische Anfälle, die nur in Teilen des Gehirns oder in bestimmten Hirnregionen auftreten, werden als fokale Anfälle bezeichnet. Sie gehen immer von einem bestimmten Bereich des Gehirns aus und betreffen auch im weiteren Verlauf meist nur eine Gehirnhälfte. Allerdings können sie sich auch zu einem generalisierten Anfall ausweiten, also beide Hirnhälften betreffen. Man spricht dann von einem fokal zu bilateral (beidseitig) tonisch-klonischen Anfall (in aller Regel verbunden mit einem Bewusstseinsverlust; früher: sekundär generalisierter Anfall). Deshalb werden fokale Anfälle teils auch als epileptische Anfälle mit fokalem Beginn bezeichnet.
Erstmalige und nicht akut-symptomatische Anfälle sind bei Erwachsenen zu ca. zwei Dritteln fokale Anfälle.1 Demzufolge treten fokale Epilepsien, die lediglich Teile des Gehirns betreffen bei Erwachsenen, also weitaus häufiger auf als primär generalisierte Epilepsien, die beide Hirnhälften betreffen.
Sie sind in der Regel gut therapierbar: Bei etwa 60 bis 70 Prozent der Patientinnen und Patienten führt die Behandlung einer fokalen Epilepsie mit Anfallssuppressiva zur Anfallsfreiheit.2,3
Man unterscheidet folgende Arten von fokalen Anfällen:
Bei einem komplex-fokalen Anfall nehmen Patientinnen und Patienten den epileptischen Anfall nicht bewusst wahr und können sich später nicht daran erinnern. Dies ist die bei Erwachsenen am häufigsten beobachtete Anfallsform.
Während eines nicht bewusst erlebten fokalen Anfalls ist eine direkte Ansprache der Betroffenen nur bedingt oder gar nicht möglich. Begleitend zum Anfall können dennoch komplexe Handlungsabläufe durchgeführt werden. So kann es beispielsweise vorkommen, dass Betroffene plötzlich aufspringen und einen Tisch quer durch das Zimmer schieben. Trotz der aktiven Handlung ist das Bewusstsein während des Anfalls aber mehr oder weniger eingeschränkt.
Die bewusst erlebten fokalen Anfälle treten in unterschiedlichen Formen auf:
Ein bewusst erlebter fokaler Anfall, der nur wenige Sekunden andauert, wird als Aura bezeichnet. Zum einen können Auren häufig in einen fokalen Anfall mit Bewusstseinsstörung übergehen. Zum anderen können Auren einem Anfall vorangehen, der sich weiter auf beide Gehirnhälften ausweitet.).
Bei bewusst erlebten fokalen Anfällen sind Betroffene häufig ansprechbar und erleben den epileptischen Anfall mit. Sie können aber nichts gegen die Symptome unternehmen, wie z. B. gegen das Zucken der Hand.
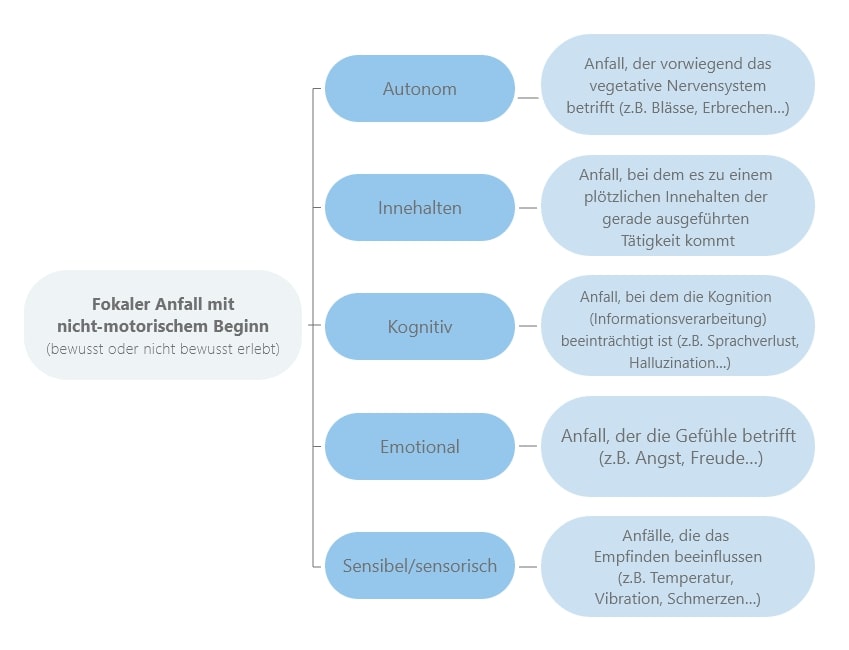
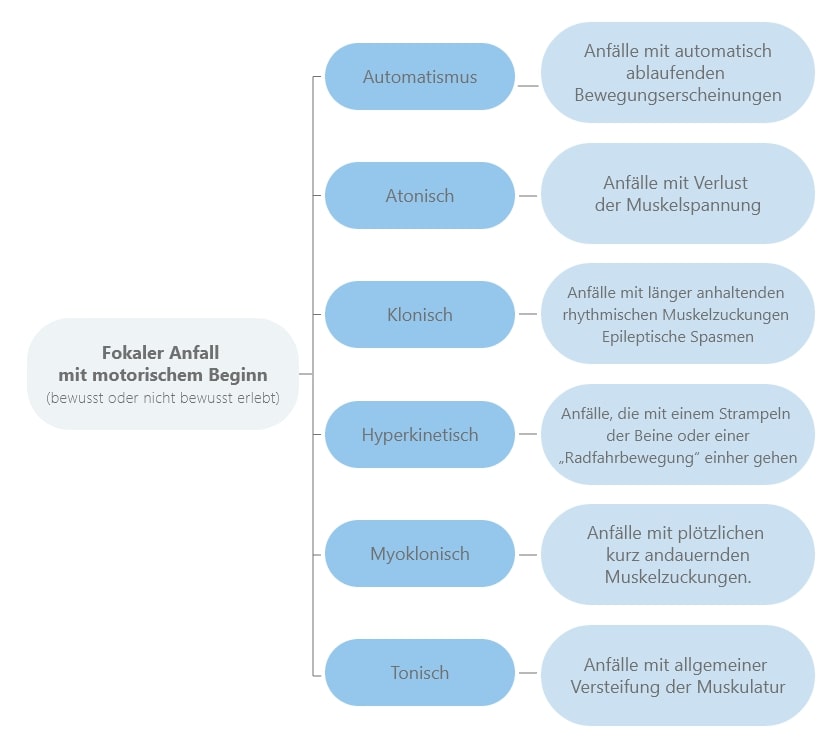
Die Symptome fokaler Anfälle richten sich nach dem Ursprungsort im Gehirn.
Fokale Anfälle mit nicht-motorischem Beginn äußern sich durch Handlungen, die nicht durch typische Bewegungsmuster gekennzeichnet sind, sondern vielmehr durch Veränderungen von Gefühlen oder anderen Körpervorgängen wie:
Auch die Sinneswahrnehmung der Patientinnen und Patienten kann gestört werden. So können Sehen, Hören, Schmecken, Riechen oder Tasten beeinträchtigt werden. Beispielsweise könnten Betroffene Blitze sehen, Objekte im Raum kleiner oder größer wahrnehmen als sie eigentlich sind, Geräusche oder Stimmen hören, einen komischen Geschmack im Mund wahrnehmen, etwas Merkwürdiges riechen sowie unter Temperatur-Missempfindungen, Kribbeln oder Lähmungserscheinungen leiden.
Fokale Anfälle mit motorischem Beginn äußern sich durch unfreiwillige Veränderungen der Muskelbewegung (Motorik).
Es gibt dabei verschiedene motorische Symptome:
Ebenso können fokale Epilepsien mit nicht-motorischen Symptomen einhergehen, bzw. mit solchen beginnen, bevor dann teils im weiteren Verlauf motorische Symptome wie eine Myoklonie, Klonie oder Atonie hinzukommen können.
Typische nicht-motorische Symptome:
Manche Epilepsie-Patientinnen und Patienten wiederholen ständig bestimmte Handlungsmuster. Dafür gibt es einen medizinischen Begriff: Die sogenannten Automatismen prägen häufig nicht bewusste erlebte fokale Anfälle. Die Bewegungen sehen dabei zwar aus wie ganz normale alltägliche Handlungen, werden aber von den Betroffenen nicht willkürlich gesteuert. Typische Automatismen sind beispielsweise ganz normale Laufbewegungen oder das Nicken mit dem Kopf. Es kann aber auch sein, dass Betroffene einfach mit der Bewegung bzw. Tätigkeit fortfahren, die sie schon vor dem Anfall ausgeübt haben.
Typische Automatismen bei nicht nicht bewusst erlebten fokalen Anfällen:
Wichtiger Hinweis: Wir sind rechtlich dazu verpflichtet, folgende Informationen ausschließlich Ärztinnen und Ärzte bzw. Menschen mit Gesundheitsberufen zur Verfügung zu stellen. Deshalb sind die Fachartikel rund um Epilepsie ausschließlich mit einem Log-in aufrufbar, z. B. via DocCheck.

Wie lautet das richtige diagnostische Vorgehen und welche Rolle spielen dabei (Fremd)-Anamnese, EEG und Bildgebung? Prof. Dr. med. Hajo Hamer gibt Ihnen in diesem interaktiven eLearning einen Überblick zu Differentialdiagnose und Therapie.

Von der Semiologie zur Diagnose
Häufig ist es für Ärztinnen und Ärzte nicht immer einfach einen epileptischen Anfall eindeutig zu diagnostizieren und zu therapieren. In dieser Online-Fortbildung gibt Ihnen Prof. Dr. med. Jörg Wellmer mittels eines interaktiven eLearnings Hilfestellungen.
Auren können nach ihrem Erscheinungsbild näher bezeichnet werden.
Dieser fokale Anfall ist für Betroffene häufig schwer in Worte zu fassen. Das Gefühl steigt aus der Magengegend als ein Druckgefühl bis zum Hals auf.
Diese Auren gehen mit Veränderungen des Geruchssinns einher und werden meistens als unangenehm empfunden.
Betroffene erleben einen veränderten Geschmack im Mund, der häufig als „metallisch“ beschrieben wird.
Diese Auren spielen sich auf der Gefühlsebene ab. Es kann zu verstärkter Angst kommen, aber auch zu erhöhten Glücksgefühlen. Die Veränderungen können außerdem als vertraut („deja vu“) oder merkwürdig fremd („jamais vu“) wahrgenommen werden. Dies wirkt sich teilweise direkt auf gedankliche Vorgänge oder sogar die Zeitwahrnehmung aus.
Hier verändert sich der Gehörsinn. Es kann zu akustischen Halluzinationen kommen, die sich von einzelnen Tönen und Geräuschen, bis hin zu Melodien entwickeln können.
Typisch für eine visuelle Aura ist eine veränderte optische Wahrnehmung, z. B. das Empfinden von Verzerrtheit, Lichtblitzen, bunten Punkte oder eines eingeschränkten Gesichtsfeldes.
Es kommt zu seltsamen Körperwahrnehmungen, z. B. Kribbeln und Ameisenlaufen.
Die folgende Tabelle gibt eine Übersicht, bei welchen Epilepsien bzw. Epilepsiesyndromen und in welchem Alter fokale Anfälle auftreten können.
| Epilepsie | In welchem Alter tritt sie auf | Typische Anfälle/Auren/Häufigkeit |
|---|---|---|
| Rolando-Epilepsie | Kinder (3 bis 12 Jahre) | Somatosensible Aura/Gefühlsstörungen, Speichelfluss, Muskelzuckungen auf einer Gesichtshälfte. Häufigste Epilepsie im Kindesalter (ca. 15%).4 Tonische Gesichtskrämpfe beginnen häufig im Schlaf. Sprachunfähigkeit kann auch postiktal (nach dem Anfall) bestehen bleiben. Mildere Bewusstseinseinschränkungen können auch interiktal (zwischen zwei Anfällen) auftreten.5 |
| Primäre Lese-Epilepsie | Jugendliche | Verkrampfungen oder Zuckungen des Kiefers, der Zunge oder der Schlundmuskulatur beim Lesen – liest der/die Betroffene trotzdem weiter, kann sich ein sekundär-generalisierter Anfall entwickeln. |
| Temporallappen-Epilepsie | Kinder, Jugendliche und Erwachsene | Bewusst erlebte fokale Anfälle, die sich zu nicht bewusst erlebten fokalen Anfällen entwicklen können, die epigastrische (auf den Oberbauch bezogene) Aura ist typisch. Es ist die häufigste Epilepsieform (ca. 40 % aller Epilepsien und 70 % aller fokalen Epilepsien).6 Ursache ist in vielen Fällen eine sogenannte Hippokampussklerose (Ammonshornsklerose; Signalveränderungen des Hippocampus). Diese Veränderungen der Hirnstruktur können durch eine Enzephalitis (Gehirnentzündung), Entwicklungsstörung, neurodegenerative Erkrankungen oder Tumore verursacht werden. Es handelt sich dann um eine strukturelle oder immunologische Epilepsie. |
| Frontalllappen-Epilepsie | Kinder, Jugendliche und Erwachsene | Kurze bewusst und nicht bewusst erlebte Anfälle in hoher Frequenz, denen meistens eine Aura vorausgeht. Häufig motorische Symptome wie Strampeln oder Um-sich-Schlagen. Der Kopf wird oft vom Epilepsie-Herd weggedreht, blickt also zur gesunden Seite/Hirnregion. Auch eine Tonisierung (erhöhte Muskelspannung), Speichelfluss, Sprachhemmungen und Aufmerksamkeitsstörungen (Pseudo-Absencen) können auftreten. Etwa 10 % aller fokalen Epilepsien sind Frontallappen-Epilepsien. |
| Parietallappen-Epilepsie | Kinder, Jugendliche und Erwachsene | Seltene Form der Epilepsie mit bewusst erlebten fokalen Anfällen, die vor allem durch sensorische Symptome geprägt werden, wie z. B. Schwindel, Ameisenlaufen, Taubheitsgefühle und Halluzinationen (vor allem akustische). Zusätzlich kann sich ein solcher Anfall durch den sogenannten "march of convulsion" (Ausbreitung motorischer und sensibler Symptome von außen nach innen) zu einem Jackson-Anfall entwickeln. Jeder fokale Anfall, der sich durch den "march of convulsion" ausbreitet, wird, unabhängig von seinem eigentlichen Herd/Ursprung als Jackson-Anfall bezeichnet. Auch ein Übergang der Parietallappen-Epilepsie hin zu temporalen und frontalen Anfällen ist möglich, die dann also den Symptomen einer Temporallappen- oder Frontallappen-Epilepsie ähneln.7 |
| Okzipitallappen-Epilepsie | Kinder, Jugendliche und Erwachsene | Häufiger sind hier generalisierte Anfälle. Aber auch bewusst erlebte fokale Anfälle sind möglich, die sich schnell zu sekundär-generalisierten Anfällen (auf weitere Hirnregionen) ausweiten. Bei fokalen Anfällen dieser Art sind optische Halluzinationen bzw. eine Wahrnehmung von Blitzen oder Lichtpunkten typisch. Die Okzipitallappen-Epilepsie entwickelt sich häufig zu temporalen und frontalen Anfällen weiter.5 |
Was unterscheidet einen generalisierten von einem fokalen Anfall? Im Gegensatz zu fokalen Anfällen sind bei generalisierten Anfällen immer beide Gehirnhälften betroffen. Fast alle Formen der „generalisierten Anfälle“ beinhalten den Verlust des Bewusstseins. Eine Ausnahme sind in diesem Fall die myoklonischen Anfälle (ruckartigen Zuckungen), bei denen das Bewusstsein meist erhalten bleibt. Typisch für generalisierte Epilepsien sind Krampfanfälle, also Anfälle mit motorischen (die Bewegung betreffenden) Symptomen. Sie treten bei generalisierten Anfällen häufiger auf als bei fokalen Anfällen.
Typische motorische Symptome:
Typische nicht-motorische Symptome:
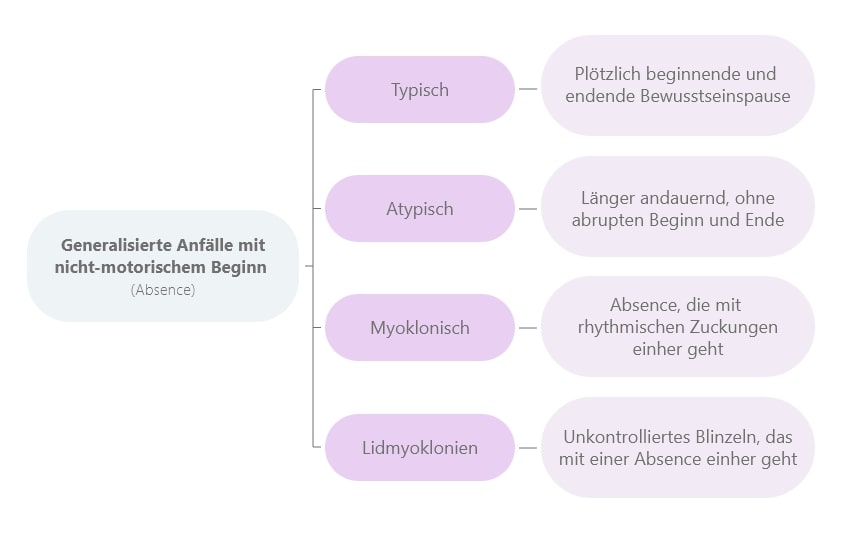
Nicht-motorische Symptome äußern sich bei generalisierten Anfällen primär in Form von Absencen, die wie folgt unterteilt werden:
Während eines Anfalls kann die Ausbreitung unterschiedlich verlaufen und das gesamte Hirnareal betreffen. Generalisierte epileptische Anfälle treten in verschiedenen Unterformen auf.
Wie fokale Anfälle werden auch Anfälle einer generalisierten Epilepsie in zwei Kategorien unterteilt:
Die meisten generalisierten Anfälle gehen mit einer Bewusstseinsstörung einher, weshalb hier nicht – wie bei den fokalen Anfällen - zwischen bewusst- und nicht bewusst-erlebten Anfällen unterschieden wird.
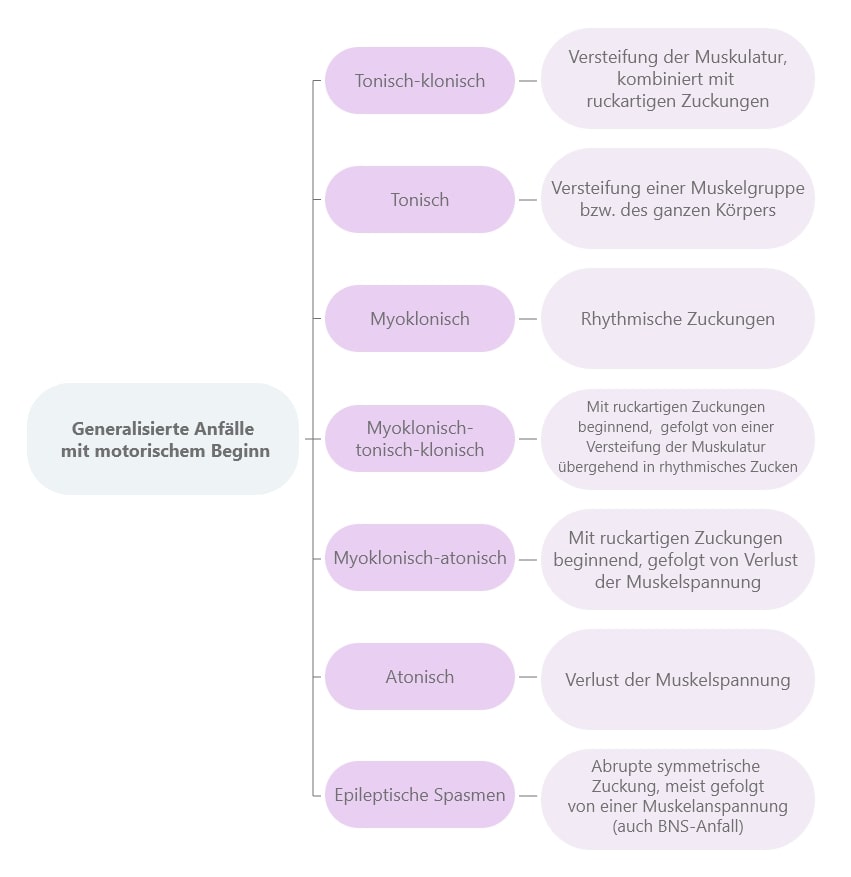
Treten keine Bewusstseinsstörungen auf, sondern äußert sich der Anfall mit schnellen und ruckartigen Muskelzuckungen? Dann handelt es sich um einen myoklonischen Anfall. Dieser betrifft häufig Kopf und Arme, es können aber auch die Beine beteiligt sein.
Bei einem klonischen Anfall treten länger anhaltende rhythmische Zuckungen auf.
Tonische Anfälle sind gekennzeichnet durch eine andauernde Muskelanspannungen, die länger anhalten kann. Meist dauert ein solcher epileptischer Anfall aber nur wenige Sekunden und er tritt oft im Schlaf auf. Säuglingen und Kindern sind öfter betroffen als Erwachsene. Geht der tonische Anfall in einen klonischen Anfall über, dann spricht man von einem tonisch-klonischen Anfall, dem sog. "Grand-mal-Anfall".
Der tonisch-klonische Anfall oder auch Grand-mal-Anfall ist die am häufigsten mit der Krankheit Epilepsie in Verbindung gebrachte Anfallsform.
Die Symptome dieses epileptischen Anfalls äußern sich meist in einem initialen Schrei des/der Patient*in. Auch von einem Stöhnen berichten Angehörige häufig. Anschließend kommt es zum Bewusstseinsverlust, weshalb auch akute Sturzgefahr zu Beginn eines tonisch-klonischen Anfalls besteht. Dem folgt eine Anspannung der gesamten Körpermuskulatur (tonische Phase), die dann in Zuckungen des Körpers übergeht (klonische Phase). Es kann auch zu vermehrtem Speichelfluss bzw. Schaum vor dem Mund kommen.
Aufgrund des Bewusstseinsverlustes können sich Patientinnen und Patienten im Nachhinein nicht mehr an den Anfall erinnern kann, es kommt also zu einer Amnesie für die Dauer des Anfalls. Auch die Blaufärbung der Lippen (Zyanose) ist ein typisches Epilepsie-Symptom, wenn es in Zusammenhang mit einem Grand-mal-Anfall auftritt. Die Blaufärbung entsteht durch die Verkrampfung der Atemmuskulatur während des Anfalls, so dass Betroffene keine Luft bekommen. Der Atemstillstand (Apnoe) kann bis zu 30 Sekunden andauern, führt aber nicht zum Ersticken der Patientinnen und Patienten. Im Anschluss an einen Grand-mal-Anfall sind die meisten Patientinnen und Patienten sehr erschöpft und müssen sich erholen. Diese Phase der Reorientierung kann ein bis zwei Stunden dauern. Hier kann es gelegentlich zum sog. postiktalen Schlaf kommen.
Die klinische Untersuchung zeigt außerdem folgende Symptome eines Grand-mal-Anfalls:
Ein Grand-mal-Anfall verläuft in zwei Phasen:
1. Phase – tonisch:
2. Phase – klonisch:
Nach dem Anfall
Verliert man die Muskelkraft, spricht man von einem atonischen Anfall. Diese epileptischen Anfälle resultieren häufig in Stürzen.
| Epilepsie | In welchem Alter tritt sie auf | Typische Anfälle / Häufigkeit |
|---|---|---|
| Lennox-Gastraut-Syndrom | Kinder (2 bis 6 Jahre) | Typisch sind mehrere Anfälle pro Tag, wobei keine andere Form der Epilepsie eine so große Vielfalt der Anfallsformen aufweist. Am häufigsten sind tonische Anfälle, die bei 90% der betroffenen Kinder im Schlaf auftreten.8,9 Auch myoklonische Anfälle sind häufig, vor allem wenn das Kind müde ist. Doch auch alle anderen Anfallsformen wie atonische Anfälle, atypische Absencen und auch Grand-mal-Anfälle können vorkommen. Außerdem kann das Syndrom oft zu einem Status epilepticus (SE) führen |
| Dravet-Syndrom (SMEI) | Babys / Kleinkinder (3. bis 12. Lebensmonat) | Eine sehr seltene Form der Epilepsie, die bei Jungen häufiger als bei Mädchen auftritt und die geistige Entwicklung beeinträchtigen kann. Sie wird auch als schwere frühkindliche Grand-mal-Epilepsie oder schwere myoklonische Epilepsie im Säuglings- und Kleinkindalter (SMEI = Severe Myoclonic Epilepsy in Infancy) bezeichnet. Symptomatisch äußert sich das Dravet-Syndrom unter anderem durch heftige rhythmische Zuckungen der Muskeln von Armen, Beinen und Gesicht sowie durch Versteifungen der Glieder und des Rumpfes, gepaart mit Bewusstlosigkeit. Die Anfälle wiederholen sich ungewöhnlich rasch und können in einen Status epilepticus münden. |
| Kindliche Absence-Epilepsie (CAE = Childhood Absence Epilepsy) | Kinder (4 bis 10 Jahre) | Kurze und eher unkomplizierte Absencen, die für fünf bis zwanzig Sekunden anhalten. Typisch sind kurze Phasen des Starrens, die unvermittelt während einer Aktivität auftreten. Begleiterscheinungen können Schmatzen, Seufzen oder das Flattern der Augenlider sein. Auch leichte Zuckungen des Kopfes sind möglich. |
| Juvenile Absence-Epilepsie (JAE) | Jugendliche | Es kommt häufig zu sporadisch auftretenden Absencen, die aber auch mit generalisierten tonisch-klonischen Anfällen und myoklonischen Anfällen einhergehen können, allerdings eher selten. Im Verlauf der Erkrankung kann sich außerdem eine Aufwach-Grand-mal oder eine juvenile myklonische Epilepsie entwickeln. |
| Juvenile myoklonische Epilepsie (JME) | Jugendliche (10 bis 18 Jahre) und Erwachsene | Die JME tritt meist zwischen dem 10. und 18. Lebensjahr erstmalig auf. Typisch sind myoklonische Zuckungen (Patient*in ist bei Bewusstsein), die arhythmisch vor allem in Schultern und Armen auftreten. Es kommt auch zum „Wegschleudern“ der Extremitäten, ähnlich wie bei einem elektrischen Schlag. Die Myklonien ohne, oder nur mit leichter Bewusstseinsstörung, können sich zu tonisch-klonischen Anfällen mit Bewusstseinsstörung entwickeln. Meistens treten die Anfälle nach dem Aufstehen auf, vor allem wenn der/die Betroffene unvermittelt geweckt wird. Wenig Schlaf, Alkoholkonsum und die sogenannte „Flickerstimulation“ (Auslöser wie Stroboskoplicht) erhöhen die Anfallswahrscheinlichkeit. Im weiteren Verlauf der Epilepsie kann es auch zu Grand-mal-Anfällen kommen. |
| Aufwach-Grand-mal | Jugendliche (15 bis 17 Jahre) | Bei dieser Form der Epilepsie kommt es innerhalb kurzer Zeit nach dem Aufstehen zu tonisch-klonischen Anfällen. Bei Entspannung kann es ebenfalls zu Anfällen kommen, weshalb diese auch als sogenannte Feierabend-Anfälle bezeichnet werden. Die Anfälle treten eher selten auf und sind mit einer individuellen Medikation typischer Anfallssuppressiva gut in den Griff zu bekommen. |
| West-Syndrom (BNS-Epilepsie) | Babys und Kleinkinder (3. bis 12. Lebensmonat) | Diese Form der Epilepsie ist altersgebunden und hängt ursächlich stark mit sehr frühen organischen Hirnschäden oder Auswirkungen anderer Erkrankungen auf das Gehirn zusammen. Die starke Altersgebundenheit ist markant dafür, dass diese Epilepsie eng mit dem Reifungszustand des Gehirns verknüpft ist. Typisch sind die drei unterschiedlichen Anfallsformen Blitz-Nick-Salaam, weshalb die Erkrankung auch als BNS-Epilepsie bezeichnet wird. Blitz-Anfälle sind heftige myoklonische Zuckungen, die den gesamten Körper oder einzelne Körperregionen durchziehen, aber nur für Sekundenbruchteile. Auffällig ist vor allem die Beugehaltung der Beine, während auch generell eher Beuge- als Streckmuster typisch sind. Nick-Anfälle bezeichnen Zuckungen der Muskulatur des Nackens und des Halses, wobei das Kinn zur Brust gebeugt oder der Kopf ruckartig eingezogen wird, was einem Nicken ähnelt. Bei den Salaam-Anfällen bewegen sich Kopf und Rumpf ruckartig nach vorne. Die Arme werden nach oben geworfen und wieder gebeugt, wobei es auch zu einem Zusammenführen der Hände vor der Brust kommen kann. In langsamer Abfolge ähneln diese Bewegungen dem orientalischen Friedensgruß „Salaam“, wonach die Anfälle benannt wurden. Die Anfälle treten häufig nach dem Aufstehen und vor dem Schlafengehen auf, können aber auch während des Schlafens einsetzen und zum Aufwachen führen. |
| Doose-Syndrom (MAE = Myoklonisch Astatische Epilepsie) | 2 bis 6 Jahre | Hierbei handelt es sich um eine seltene Epilepsie mit myoklonisch-astatischen Anfällen. 2-4 % der Kinder mit Epilepsie sind betroffen und Jungen in etwa doppelt so häufig wie Mädchen.10 Charakteristisch sind myoklonische und myoklonisch-atonische Anfälle. Die Anfälle dauern nur wenige Sekunden, können anfangs aber in sehr hoher Frequenz auftreten. Die atonische Komponente der Anfälle besteht aus einem plötzlichen Verlust der Muskelpannung, wodurch die Patientinnen und Patienten sofort in sich zusammenfallen. |
| Ohtahara-Syndrom | Babys (ab 10 Tagen nach der Geburt bis zum 3. Monat) | Eine äußerst seltene Form der Epilepsie. Sie tritt bei Neugeborenen bereits innerhalb von 10 Tagen bis 3 Monaten nach der Geburt auf. Am typischsten sind tonische Anfälle, die bis zu einer Minute anhalten. Doch auch atypische Absencen, komplex-fokale Anfälle und myoklonische Anfälle können auftreten. Ein häufiges Anzeichen ist die Muskelhypotonie, eine Verringerung der Muskelspannung, wodurch die Säuglinge ihren Kopf nicht dem Alter entsprechend selbst aufrechthalten können. |
Bei Absence-Epilepsien kommt es zu einer plötzlichen Bewusstseinsstörung, so dass die Betroffenen ihre momentane Tätigkeit für die Dauer des Anfalls plötzlich unterbrechen. Es handelt sich hierbei um nicht-motorische generalisierte Anfälle, die als mildere Form einer generalisierten Epilepsie betrachtet werden. Es kommt nämlich nicht zum Verkrampfen oder zum Sturz, sondern zu einem geistigen Aussetzer bei den Betroffenen. Deshalb wurden Absencen früher auch als „Petit mal“ bezeichnet, was man auch als „das kleine Übel“ übersetzen könnte (im Vergleich zum motorischen "Grand mal"-Anfall, dem "großen Übel").
Die Betroffenen starren bei dieser Form eines epileptischen Anfalls oft ins Leere und haben ein ausdrucksloses Gesicht. Die Anfälle können mehrere Sekunden dauern und sich stark gehäuft über den Tag wiederholen.
Absence-Epilepsien gehen, im Gegensatz zu vielen anderen epileptischen Anfällen, meist keine Aura und kein Prodrom voraus. Die Patientinnen und Patienten können sich an den Anfall nicht erinnern und fahren mit ihrer Tätigkeit nach dem Anfall wieder fort.
Obwohl diese Anzeichen typisch für Absencen sind, werden sie von Laien vielfach nicht als Symptome einer Epilepsie erkannt. Absence-Epilepsien sind eine häufige Form der generalisierten Epilepsien des Kindesalters und werden zunächst meist als Unkonzentriertheit oder Träumerei missinterpretiert.
Sie werden mit antiepileptischen Medikamenten behandelt, wodurch Eltern und Kinder die Anfälle meist gut unter Kontrolle bekommen. Die Prognose ist also günstig. Bis zu 80 % der Betroffenen werden durch eine Therapie anfallsfrei. Es kann zu wenigen Anfällen innerhalb eines Jahres bis hin zu mehreren hundert am Tag kommen. Wie oft epileptische Anfälle dieser Art auftreten, hängt vom individuellen Krankheitsbild ab. Bei Erwachsenen und Jugendlichen sind Absence-Epilepsien zwar bekannt, jedoch treten sie weitaus häufiger im Kindesalter auf. Dabei sind Mädchen deutlich öfter betroffen als Jungen.
Absencen können weiter in Kategorien unterteilt werden:
Typische Absencen:
Atypische Absencen:
Während des Verlusts des Bewusstseins kommt es zu leichten Myoklonien (Rhythmischen Zuckungen). Davon können auch andere Teile des Gesichts, des Halses, der Schultern oder der Arme betroffen sein. Geht die Absence mit Myoklonien des Augenlids einher, spricht man von Lidmyoklonien.
Fortbildungen aus den Bereichen der Neurologie und Neuropädiatrie.


Wie wird Epilepsie richtig behandelt?
Der Einsatz von Anfallssuppressiva zur Epilepsie-Behandlung ist wichtig. Doch auch die Ernährung und die sog. Vagusnerv-Stimulation spielen eine Rolle. Lesen Sie hier mehr darüber.
Anfallskalender als PDF herunterladen
Ein Anfallskalender kann helfen, Anfälle festzuhalten und für den Arzt zu protokollieren
Autor: Desitin Arzneimittel GmbH
Wichtiger Hinweis: Wir sind rechtlich dazu verpflichtet, folgende Informationen ausschließlich Ärztinnen und Ärzte bzw. Menschen mit Gesundheitsberufen zur Verfügung zu stellen. Deshalb sind die Fachartikel rund um Epilepsie ausschließlich mit einem Log-in aufrufbar, z. B. via DocCheck.

Ein einzelner epileptischer Anfall erfordert normalerweise keine medizinische Hilfe und stellt damit auch keinen medizinischen Notfall dar. Es gibt dennoch einige Maßnahmen, die man im Falle eines Falles ergreifen sollte, um dem*der Betroffenen und dem*der behandelnden Ärzt*in zu helfen. Hier haben wir die wichtigsten Punkte für Sie zusammengefasst. Angefangen von einfachen Maßnahmen, bis hin zu Notfallmedikamenten.

Wann ist ein epileptischer Anfall ein Notfall?
Generalisiert tonisch-klonische Anfälle (GTCS) sehen meist schlimm aus, aber wie sinnvoll ist tatsächlich die Verabreichung eines sedierenden Notfallmedikamentes? Auch in diesem Fall gilt, dass der Patient individuell betrachtet werden muss, z. B. ob dieser einen ersten Anfall erlitten hat oder zu Anfallsserien neigt. Prof. Elger geht auch auf die Frage ein, was bei längeren Anfällen von mehr als 5 Minuten Dauer zu tun ist.
Das Desitin Redaktionsteam besteht aus den Bereichen Medical Affairs und Product Management. Um Ihnen die besten Inhalte zu bieten, arbeiten wir zusätzlich mit Expertinnen und Experten zusammen. Das Team wird um ausgewählte Ärztinnen und Ärzte sowie Fachjournalistinnen und Fachjournalisten ergänzt. Diese schreiben regelmäßig für uns und bereichern desitin.de mit ihren fachlichen Beiträgen. Schreiben Sie uns bei Fragen auch gerne eine E-Mail an info@desitin.de.
1 Forsgren L et al. Incidence and clinical characterisation of unprovoked seizures in adults. A prospective population-based study. Epilepsia 1996;37(3):224-229. doi:10.1111/j.1528-1157.1996.tb00017.x.
2 Sander JW et al. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet 1990;336(8726):1267-1271. doi:10.1016/0140-6736(90)92959-l.
3 Brandt C. Akut-symptomatische epileptische Anfälle: Inzidenz, Prognose und Aspekte der antiepileptischen Behandlung. Aktuelle Neurologie 2012;39(09):480-485.
4 Hacke W. Neurologie. 14. Auflage, Springer Verlag 2016. ISBN: 978-3-662-46891-3.
5 Fröscher W. Die Epilepsien: Grundlagen - Klinik - Behandlung. Schattauer Verlag 2004. ISBN: 978-3-794-52131-9.
6 Neubauer BA, Groß S, Hahn A. Epilepsie im Kindes- und Jugendalter. Dtsch Arztebl 2008;105(17):319-27. doi: 10.3238/arztebl.2008.0319.
7 Schmidt D, Elger CE. Praktische Epilepsiebehandlung. 3. Auflage, Georg Thieme Verlag 2005. ISBN: 978-3-131-16823-8.
8 DocCheck. Lennox-Gastaut-Syndrom. Online verfügbar unter: https://flexikon.doccheck.com/de/Lennox-Gastaut-Syndrom. Zuletzt abgerufen: April 2021.
9 RRoger J, Dravet C, Bureau M. The Lennox-Gastaut syndrome. Cleve Clin J Med 1989;56 (Suppl Pt 2):172-180.
10Schmidt Dieter. Epilepsie. Diagnostik und Therapie für Klinik und Praxis. Schattauer Verlag 1997; S. 35. ISBN 978-3794517893.
Mehr für Epilepsie-Patienten
Um Ihnen den Alltag als Patient/in bzw. Angehörige/r zu erleichtern,
bieten wir Ihnen umfangreiche Informationen.
INFOMATERIAL
Broschüren & Downloads
PRODUKTE
Übersicht & Informationen
ZENTREN FINDER
Hilfe in Ihrer Nähe
TIPPS
Für den Alltag