

Myasthenia gravis (MG), in Deutschland häufig auch nur Myasthenie genannt, ist eine Autoimmunerkrankung. Bei solchen Erkrankungen richtet sich das körpereigene Immunsystem aufgrund einer Fehlfunktion gegen eigentlich harmlose Zellen und/oder Gewebe des Körpers und greift diese an. Je nach Art der Erkrankung entstehen so verschiedenste Symptome.

Dies kann zu Entzündungen und sogar zur Zerstörung von Gewebe und Organen führen. Das geschieht, da der Körper sogenannte Autoantikörper (AAk) bildet, die eigentlich dazu dienen, Viren oder Bakterien abzuwehren. Im Falle einer Autoimmunerkrankung richten sich diese Antikörper jedoch gegen den eigenen Körper. Bekannte Beispiele für Autoimmunerkrankungen sind Rheumatoide Arthritis, Schuppenflechte, Multiple Sklerose und Typ-1-Diabetes.
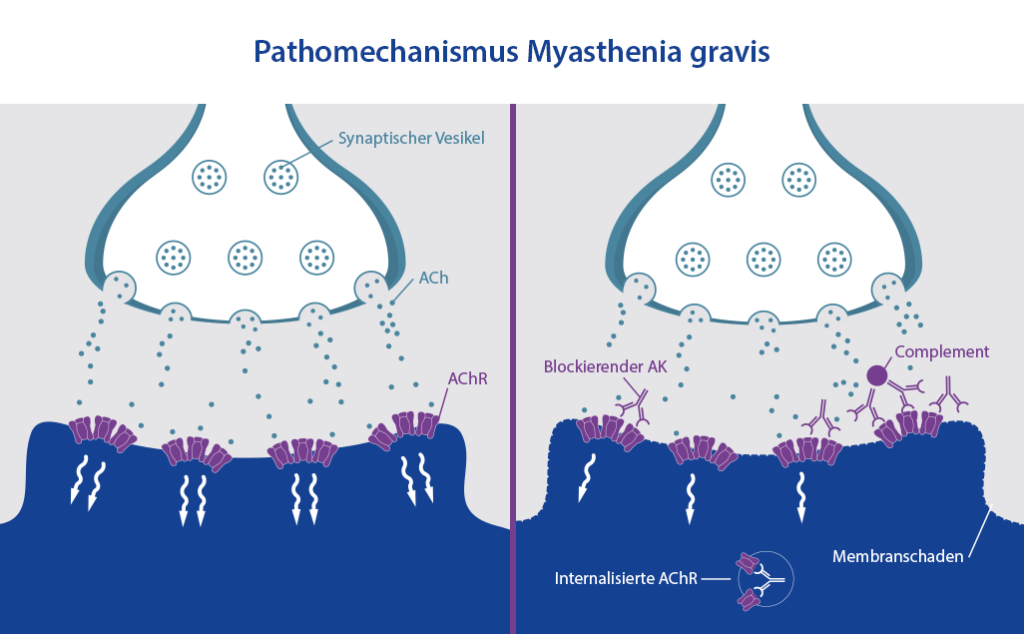
Bei Myasthenia gravis richten sich die Antikörper gegen Rezeptoren und Enzyme, welche für die Kommunikation zwischen Nervenzellen und Muskeln sehr wichtig sind. Dadurch kann es zu verschiedensten Symptomen kommen, die allesamt mit einer nachlassenden Muskelspannung zu tun haben.
Für Autoimmunerkrankungen gilt weiterhin, dass man ihnen weder effektiv vorbeugen noch sie heilen kann. Zahlreiche Therapieansätze zielen vor allem darauf ab, die Symptome deutlich zu lindern und die Lebensqualität der Betroffenen zu verbessern.
Myasthenia gravis tritt übrigens häufig bei Patientinnen und Patienten auf, die bereits an einer anderen Autoimmunerkrankung leiden, zum Beispiel an rheumatoider Arthritis, systemischem Lupus erythematodes und Erkrankungen mit Überaktivität der Schilddrüse (autoimmune Hyperthyreose), wie etwa Hashimoto-Thyreoiditis. Leiden also Menschen mit Hashimoto oder Arthritis zusätzlich an typischen Symptomen einer Myasthenia gravis, kann dies ein wichtiger Anhaltspunkt für die Diagnose der Erkrankung sein.
Die Myasthenie ist eine vergleichsweise unbekannte Erkrankung. Deshalb hat es sich DESITIN als Experte für seltene Erkrankungen und Störungen des zentralen Nervensystems, etwa Parkinson und Epilepsie, zur Aufgabe gemacht, vor allem über die Symptome der Erkrankung aufzuklären. So sollen sowohl Ärztinnen und Ärzte als auch Patientinnen und Patienten für die Diagnose der Erkrankung sensibilisiert werden. Außerdem wollen wir mit verschiedenen Produkten unseren Beitrag zur Behandlung der Myasthenia gravis leisten. Als seltene Erkrankungen (rare diseases) gelten in der EU übrigens Krankheiten, von der nicht mehr als 500 von 1.000.000 Menschen betroffen sind, also 0,05% der Bevölkerung.
Myasthenia gravis führt bei Betroffenen zu einer Störung der Kommunikation zwischen Nerven und Muskeln. Genauer gesagt wird die Reizübertragung gestört, die normalerweise die Muskelspannung aktiviert und aufrechterhält (neuromuskuläre Übertragung). Das führt zu einer sogenannten belastungsabhängigen schweren Muskelschwäche. Diese Bezeichnung wurde gewählt, da die Symptome, zum Beispiel hängende Augenlider bzw. unterschiedlich weit geöffnete Augen, sich bei Müdigkeit und schwerer Belastung intensivieren, aber nach erholsamen Phasen auch wieder abmildern können.
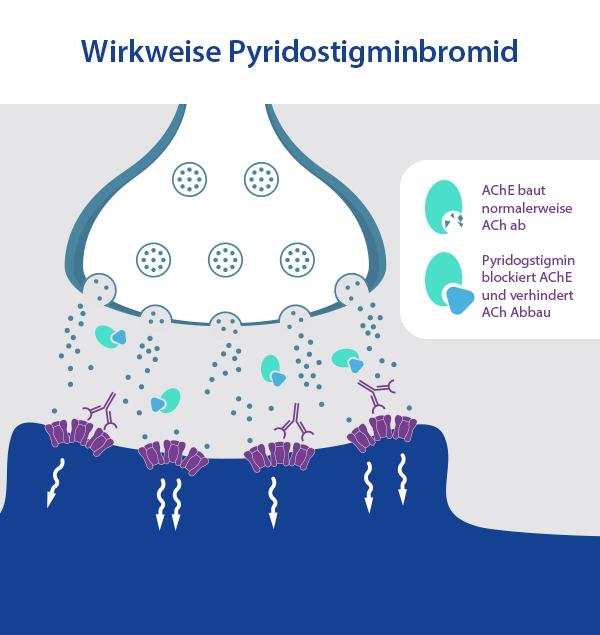
Damit unsere Nerven mit unseren Muskeln kommunizieren können, um dort Muskelkontraktionen auszulösen und aufrechtzuerhalten, müssen sie chemische Botenstoffe freisetzen, sogenannte Neurotransmitter. Für die neuromuskuläre Übertragung ist vor allem der Botenstoff Acetylcholin (ACh) entscheidend. An der neuromuskulären Verbindungstelle, also den Rezeptoren der Muskeln, docken diese an und stimulieren die Muskulatur, welche sich dann zusammenzieht. Diese Rezeptoren sitzen auf der motorischen Endplatte. Nach der Kontraktion wird das ACh von einem Enzym, der Cholinesterase, abgebaut und die Nervenenden nehmen es wieder auf. Es wird dann erneut gespeichert und steht nach einiger Zeit wieder zur Verfügung.
Bei der häufigsten Form der Myasthenie bildet das Immunsystem Antikörper gegen einen bestimmten Rezeptor der Muskeln, der auf den Neurotransmitter Acetylcholin reagiert (ACh-Rezeptoren), wodurch die Kommunikation zwischen Nervenzellen und Muskeln gestört wird, da das ACh nicht mehr „andocken“ kann. Warum genau der Körper seine eigenen Acetylcholinrezeptoren angreift, ist, wie bei so vielen Autoimmunerkrankungen, nicht abschließend geklärt. Eine besondere Rolle wird jedoch einer Fehlfunktion des Thymus zugeschrieben, worüber Sie mit einem Klick auf den Link mehr erfahren. Zudem gibt es einige Betroffene, die keine Antikörper gegen Acetylcholinrezeptoren aufweisen, sondern gegen ein Enzym, welches an der Bildung und Regeneration der neuromuskulären Verbindungsstelle beteiligt ist. Diese Form der Myasthenie muss anders behandelt werden.

Informationen für Ärztinnen und Ärzte
Fachinformationen, Servicematerialien und
vieles mehr zum Thema Myasthenia gravis
Es gibt verschiedene Erkrankungen, die eine Störung dieser Art der Übertragung von Nervenimpulsen auf die Muskeln verursachen können. Die häufigste dieser Erkrankungen ist die Myasthenia gravis, seltener tritt das Lambert Eaton-Syndrom auf und es gibt auch sehr seltene angeborene myasthene Syndrome.
Da es sich um eine Autoimmunerkrankung handelt, kommen verschiedene Auslöser infrage, welche eine Myasthenia gravis verursachen oder ihre Ausprägung verschlimmern können, bis hin zu einem Schub (myasthene Krise), den ca. 10-15 % aller Patientinnen und Patienten mit Myasthenie mindestens einmal im Leben erleiden. Auslöser eines solchen Schubs sind oft Infektionskrankheiten. Generell kommen aber verschiedene Trigger infrage, die alle eines gemeinsam haben; sie beeinflussen das Immunsystem, welches maßgeblich am Verlauf einer Autoimmunerkrankung beteiligt ist.
Mit etwa einem Erkrankten auf 10.000 Personen zählt die Myasthenia gravis zu den seltenen Erkrankungen innerhalb der EU. Im Allgemeinen ist die Erkrankung nicht ansteckend und ein familiäres Auftreten ist extrem selten. Ca. 10 % der Betroffenen mit der Diagnose Myasthenia gravis sind unter 16 Jahre alt.
Dabei gibt es zwei Erkrankungspeaks:
Weitere Informationen finden Sie in der Tabelle zu den verschiedenen Formen der Myasthenia gravis.
Betroffen sind also vor allem Frauen im Alter zwischen 20 und 40 Jahren, während sich die Erkrankung bei Männern eher im höheren Alter zwischen dem 50. Und 80. Lebensjahr manifestiert. Prinzipiell kann die Autoimmunerkrankung jedoch bei beiden Geschlechtern in jedwedem Alter auftreten. Besonders selten sind hingegen Säuglinge und Kinder betroffen.

Eine seltene Form der Myasthenia gravis ist die neonatale Myasthenie, die bei etwa 12 Prozent der Kinder von Müttern mit Myasthenia gravis auftritt. Da die Antikörper, die die Erkrankung verursachen, im Blut zirkulieren, können sie über die Plazenta an das ungeborene Kind weitergegeben werden. Dies führt zu Muskelschwäche beim Neugeborenen, die jedoch in den ersten Tagen bis Wochen nach der Geburt wieder verschwindet. Die übrigen 88 Prozent der Neugeborenen von Eltern mit entsprechendem Erbgut sind von dieser Form der Myasthenie nicht betroffen.
Die Antikörper, welche die Kommunikation zwischen Nerven und Muskeln stören, werden zu einem großen Teil im sog. Thymus gebildet, der hinter dem Brustbein liegt. Zudem ist es so, dass die Thymusdrüse bei vielen Patientinnen und Patienten mit Myasthenie vergrößert und übermäßig aktiv ist. Daher führt die Entfernung der Thymusdrüse in einigen Fällen zur Besserung der Myasthenie Symptome. Es ist jedoch zu beachten, dass eine vergrößerte Thymusdrüse auch ein Anzeichen für einen Thymus-Tumor (Thymom) sein kann und in diesem Fall eine Operation unerlässlich ist. Gleichzeitig können Thymome somit auch für entsprechende Veränderungen der Thymusdrüse verantwortlich sein, die in der Folge zu Myasthenia gravis führen. Bei Patientinnen und Patienten mit einer Myasthenie findet sich bei 10 – 15 % der Betroffenen ein Thymom. Doch gleichzeitig wird bei ca. 45 % aller Patientinnen und Patienten mit einem Thymom eine Myasthenie diagnostiziert.
Teilweise wird auch eine Entfernung des Thymus erwogen, obwohl kein Tumor nachweisbar ist. Das Haupt-Kriterium dafür ist das Bestehen einer generalisierten Myasthenie.








Ein großes Problem bei der Diagnose der Erkrankung ist, dass die Symptome dem Krankheitsbild häufig nicht richtig bzw. frühzeitig zugeordnet werden. Deshalb ist es wichtig, dass sowohl Ärztinnen und Ärzte als auch Angehörige und Betroffene besser darüber informiert werden, welche – oft unspezifischen - Anzeichen auf eine Myasthenie hinweisen können. Kennzeichen für eine Myasthenia gravis sind oft weniger die Anzeichen wie Müdigkeit oder erschöpfte Muskeln, sondern die Reihenfolge und Zeitpunkte ihres Auftretens.
Die Symptome selbst sind sehr vielseitig und häufig auch typisch für andere Erkrankungen. Zum Beispiel kann eine Myasthenie zu Sprechstörungen und Schluckbeschwerden führen, welche, insbesondere bei älteren Menschen, häufig den typischen Parkinson-Symptomen zugeordnet werden. Weit davor kommt es aber in vielen Fällen zunächst zu Symptomen, welche die Augen betreffen. Typisch sind das hängende Augenlid (Ptose) und Sehstörungen, wie etwa Doppelbilder.
Außerdem sind die Beschwerden von Person zu Person unterschiedlich und können sich obendrein im Laufe der Zeit verändern. Das Kardinalssymptom ist viel mehr der Zeitpunkt, zu dem die Symptome auftreten bzw. sich intensivieren und wann sie nachlassen. Die Ermüdung bestimmter Muskelpartien steigt nach Stress und Anspannung, während sich die Muskelspannung und Aktivität nach Phasen der Erholung wieder verbessert.
Dabei sind bestimmte Muskelgruppen häufiger betroffen als andere. Typisch ist zum Beispiel die Ermüdung der Augenmuskulatur und der Augenlidbewegung. Auch die Mimik und die für Kauen, Sprechen und Schlucken zuständigen Muskelgruppen sind häufig (aber nicht immer) betroffen. Typisch ist auch ein allgemeines Gefühl von „Schlappheit“ in der Muskulatur.
Oft tritt die Erkrankung sehr plötzlich auf, während der Grad der Muskelschwäche sowohl nach Tagesform als auch von Person zu Person unterschiedlich ist, was die Definition allgemeiner Anhaltspunkte für die Diagnose umso schwieriger macht.
Die Muskelschwäche der Halsmuskulatur führt häufig zu Schwierigkeiten beim Schlucken und Sprechen, während die Störungen im Bereich der Augenpartie zu Sehstörungen und einem hängenden Augenlid führen können.
Die folgenden typischen Symptome einer Myasthenia gravis können sowohl gemeinsam als auch einzeln auftreten:
In manchen Fällen kann eine sehr starke Myasthenie der Atemmuskulatur zu Atemversagen führen, was sofortige medizinische Notfallversorgung erfordert.
Die Myasthenia gravis ist eine seltene Erkrankung und es ist schwierig genaue Zahlen über ihre Häufigkeit und die Prognose zu erhalten. Dank der heutigen Behandlungsmöglichkeiten ist der Verlauf der Erkrankung in der Regel günstig und führt nicht mehr zu einer Verkürzung der Lebenserwartung. Die meisten Patientinnen und Patienten können trotz Einschränkungen ihrer körperlichen Belastbarkeit ein weitgehend normales Leben führen und ihren Beruf ausüben. Es gibt jedoch keine Möglichkeit, die Muskelkraft vollständig wiederherzustellen. Alle Patientinnen und Patienten müssen ihre individuellen Belastungsgrenzen herausfinden. Es besteht die Hoffnung, dass zukünftige Forschung noch bessere Behandlungsmöglichkeiten hervorbringt. In diesem Ratgeber finden Sie auch Tipps zum Alltag mit Myasthenia Gravis. Das Therapieziel ist die bestmögliche Krankheitskontrolle unter Wiederherstellung der Lebensqualität der Patientinnen und Patienten.
Sehstörungen sind in der Mehrzahl der Fälle das erste Symptom und können so im Frühstadium auf die Erkrankung hinweisen. Vor allem dann, wenn sie in der zweiten Tageshälfte, also nach mehrstündiger Arbeit und Belastung auftreten. Im Verlauf der Krankheit kommen dann meist weitere Symptome hinzu, wie Schluck- und Sprechstörungen oder eine plötzliche Schwäche der Arme und Beine, etwa beim Treppenstiegen und Aufrichten.
Sprechstörungen äußern sich zum Beispiel durch abgehacktes, stakkatoartiges, gehauchtes, unregelmäßiges, unpräzises und monotones Sprechen, während die Betroffenen Sprache aber sehr wohl richtig anwenden als auch verstehen. Das ist wichtig zu verstehen, da Sprechstörungen, gerade bei älteren Menschen, sonst schnell den Eindruck erwecken, als läge eine kognitive Störung vor, zum Beispiel Demenz.
Die Symptome nehmen in der Regel bei Anstrengung und zum Abend hin zu. Erholungsphasen können helfen, die Beschwerden wieder zu lindern. Seelische Belastungen, Schlafmangel, Alkoholkonsum, Fieber sowie grippale Infekte können die Störungen ebenfalls verstärken. Die Ausprägung und das Voranschreiten der Symptome ist bei den Betroffenen und im zeitlichen Verlauf jedoch sehr unterschiedlich. Alle haben ganz individuelle Probleme, funktionelle Einbußen und körperliche Beeinträchtigungen. Daher ist eine individuell auf Patientinnen und Patienten abgestimmte Therapie der Myasthenia gravis besonders wichtig.
Okuläre Myasthenie (OMG)
Generalisierte Myasthenie mit leichter / mittlerer / schwerer Ausprägung
Um sich einer Diagnose anzunähern, können Betroffene sich folgende Fragen stellen:
Um diese Annahmen zu überprüfen, sollten Betroffene die Symptome dringend bei Neurologinnen und Neurologen abklären lassen, welche den Verdacht mittels weiterer Diagnostik ggf. sichern oder widerlegen können.
Die Symptome einer Myasthenia gravis sind sehr unspezifisch, da Muskelschwächen quasi überall auftreten können. Zudem nehmen viele Betroffene es zunächst nicht unbedingt als Anzeichen für eine Krankheit wahr, wenn die Beine nach dem Treppensteigen erschöpft sind. Trotzdem gibt es einige Warnzeichen, bei denen Sie sofort einen Arzt aufsuchen sollten. Das ist immer dann der Fall, wenn alltägliche Muskelbewegungen plötzlich sehr schwerfallen.
Gehen Sie zum Arzt, wenn Ihnen Folgendes schwerfällt:
Eine generalisierte Form der Myasthenie kann alle Skelettmuskeln betreffen. Oft sind es die Muskeln in Armen und Beinen, sowie die Muskeln, die für Sprechen und Schlucken verantwortlich sind. Dadurch kann es zu Schwierigkeiten bei alltäglichen Aktivitäten kommen. Zum Beispiel beim Ausziehen von Kleidung über dem Kopf, sowie beim Einräumen von Geschirr in hoch hängende Küchenschränke. Gleichzeitig ist das Risiko für myasthene Krisen und eine Beeinträchtigung der Atemmuskulatur erhöht.
Die Myasthenia gravis gehört zu den neurologischen Erkrankungen, die sich kaum auf den ersten Blick diagnostizieren lassen. Die ersten Anzeichen wie Erschöpfbarkeit, Müdigkeit oder Augenprobleme sind auf den ersten Blick relativ unspezifisch, zudem gibt es verschiedene Subtypen. Eine Myasthenie wird daher meist durch fachlich versiertes Nachfragen erfahrener Neurologinnen und Neurologen erkannt.
Zur Untersuchung stehen folgende Methoden zur Verfügung:
Die klinische Anamnese bezüglich der medizinischen Vorgeschichte nimmt einen sehr wichtigen Stellenwert ein, ähnlich wie die Erfahrung der behandelnden Ärztinnen und Ärzte. Die Patientinnen und Patienten werden gezielt nach Doppelbildern, Kau- und Schluckbeschwerden, Gewichtsabnahme und der Verschlechterung der Symptomatik unter Belastung bzw. in der zweiten Tageshälfte gefragt.
Weiterhin werden verschiedene, in Abhängigkeit von den individuellen Beschwerden, Überprüfungen der Symptome vorgenommen.
Es können verschiedene Tests durchgeführt werden, wie z.B. eine neurologische Untersuchung, bei der Reflexe, Muskelkraft, Muskeltonus, Tastsinn und Sehvermögen, Koordination und Gleichgewicht überprüft werden.
Tests, die helfen können, die Diagnose zu bestätigen, sind z.B. der Eisbeuteltest, bei dem ein Beutel mit Eis auf das Augenlid gelegt wird, um eine eventuelle Besserung des müden Augenlids zu untersuchen.
Ein weiterer Test, der sich z. B. für die Überprüfung von Sehstörungen und anderen Symptomen der Augen eignet, ist der pharmakologische Test mit Edrophoniumchlorid. Dabei handelt es sich um einen kurzwirksamen Cholinesterasehemmer. Wie bereits eingangs in diesem Ratgeber erwähnt, bauen Cholinesterasen das Acetylcholin an den Rezeptoren der motorischen Endplatte ab. Wenn diese Enzyme gehemmt werden, erhöht sich dadurch folgerichtig kurzzeitig die Konzentration von ACh an den Rezeptoren der Muskeln. Die Muskelspannung steigt dadurch für kurze Zeit an. Vor dem Test werden die Patientinnen und Patienten gebeten, mehrmals die Augen zu schließen und zu öffnen. Durch diesen repetitiven Vorgang kommt es bei Patientinnen und Patienten mit Myasthenia gravis zur Ermüdung der Muskeln und die Augenlider beginnen zu hängen. Zudem können Sehstörungen wie Doppelbilder auftreten. Anschließend wird den Betroffenen eine Injektion mit Edrophoniumchlorid verabreicht. Nun sollen Sie die Augen erneut mehrmals öffnen und schließen. Sind die Symptome nun schwächer ausgeprägt als vor der Medikamentengabe, ist dies ein deutliches Anzeichen für das Vorliegen einer Myasthenia gravis, oder aber für das Lambert-Eaton-Syndrom.
Der Pyridostigmin-Test funktioniert ähnlich wie der Edrophiniumchlorif-Test, lediglich mit einem anderen Cholinesterasehemmer, nämlich mit dem namensgebenden Pyridostigmin. Der Test läuft ebenfalls ähnlich ab und wird ambulant durchgeführt. Primär wird er bei älteren Patientinnen und Patienten durchgeführt, indem 30-60 mg Pyridostigmin verabreicht werden. Allerdings nicht als Injektion, sondern oral. Bei Patientinnen und Patienten mit Myasthenia gravis wird sich die Symptomatik nach spätestens 45 bis 60 Minuten verbessern, was zu einer positiven Bewertung des Tests führt.
Blutuntersuchungen sind wichtig, um Antikörper zu erkennen, die die Rezeptoren, an denen die Nervenimpulse die Muskeln ansteuern, beeinträchtigen oder die Enzyme, welche an der Bildung der Rezeptoren beteiligt sind. Getestet wird zum Beispiel auf Anti-Acetylcholinrezeptor-AK, Anti-MuSK-AK, Anti-LRP4-AK und Anti-Titin-AK. Auf Letztere wird das Blut untersucht, wenn der Verdacht auf Tumore der Thymusdrüse besteht, sogenannte Thymome. Sowohl gutartige als auch bösartige Tumore des Thymus können Myasthenia gravis verursachen. Auf dieser Basis können auch bestimmte Behandlungsentscheidungen, zum Beispiel für die Entfernung des Thymus, getroffen werden. Davon profitieren mitunter auch Patientinnen und Patienten ohne Thymome, da die Antikörper, die sich gegen die ACh-Rezeptoren oder andere wichtige Enzyme richten, vor allem in der Thymusdrüse gebildet werden. Die Entfernung des Thymus, ohne das Vorliegen von Tumoren, wird vor allem bei Patientinnen und Patienten mit generalisierter Myasthenie in Erwägung gezogen, da bei dieser Form der Erkrankung das Risiko für myasthene Krisen erhöht ist.
Repetitive Nervenstimulation und Einzelfaser– Elektromyographie (EMG) messen die elektrische Aktivität zwischen dem Gehirn und den Muskeln. Durch die repetitive Stimulation kann eine schnelle Ermüdung der stimulierten Muskeln erkannt werden.
Eine CT oder Thorax-MRT kann durchgeführt werden, um eventuelle Tumoren oder Abnormalitäten im Thymus zu erkennen.
Lungenfunktionstests können durchgeführt werden, um zu prüfen, ob die Erkrankung Auswirkungen auf die Atmung hat. Das ist wichtig, denn wenn die Myasthenia gravis sich auf die Atemmuskulatur auswirkt, kann dies zu einer Ateminsuffizienz führen, die lebensbedrohlich ist. Störungen der Atmung bei Myasthenia gravis können sich in Form einer schwachen, heiseren Stimme, eines schwachen Hustens oder eines häufigen Atemholens beim Sprechen äußern. Diese Form der Erkrankung birgt auch ein erhöhtes Risiko für eine so genannte myasthene Krise, die durch Atemlähmungen und schwere Schluckstörungen gefährlich werden kann.
Die Diagnose der Myasthenia gravis kann umso zielführender ablaufen, je besser Patientinnen und Patienten und deren Angehörige wissen, was sie bei einem Arzttermin erwartet. Insbesondere auf bestimmte Fragen in der Anamnese kann man sich gezielt vorbereiten, woraufhin die Ärztinnen und Ärzte besser abwägen können, welche anschließenden Untersuchungen notwendig sind. Zusätzlich gibt es Fragen, die Patientinnen und Patienten Ihren Ärztinnen und Ärzte stellen können, um die Erkrankung besser zu verstehen.
Natürlich läuft jeder Termin in der Neurologie etwas anders ab, aber es gibt dennoch einige Dinge, die auf jeden Fall in der Anamnese abgefragt werden.
Diese Fragen werden häufig gestellt:
Um sich auf diese Fragen vorzubereiten, kann es helfen, dass Sie sich zunächst genau mit den möglichen Symptomen einer Myasthenie beschäftigen und im Vorfeld ein kleines Symptome-Tagebuch führen, indem Sie die Symptome und die Uhrzeit notieren, zu der sie aufgetreten sind, ebenso wie die Dinge, die Sie kurz vor dem Auftreten der Symptome gemacht haben.
Zudem kann es hilfreich sein, wenn Sie mit Freund*innen und Ihrer Familie über Ihre Symptome sprechen.
Sammeln Sie gemeinsam mit Angehörigen und Freund*innen folgende Informationen:
Außerdem gibt es einige Fragen, die Sie Ihren Ärztinnen und Ärzte gerne im Gespräch stellen können, sofern diese Punkte nicht bereits besprochen wurden:
In den überarbeiteten Leitlinien zu Myasthenia gravis wird darauf hingewiesen, dass bei Antikörper-negativer Myasthenia gravis in Abhängigkeit vom Ansprechen auf Immuntherapie als Differentialdiagnose das sogenannte kongenitale myasthene Syndrom (CMS) in Betracht gezogen werden sollte.
Kongenitale Myasthene Syndrome (CMS) sind seltene genetische Erkrankungen, die nicht auf autoimmunen, sondern genetischen Störungen der neuromuskulären Übertragung beruhen. Die Symptome beginnen in der Regel bereits bei Geburt oder in den ersten beiden Lebensjahren. Da die CMS keine autoimmunologischen Ursachen hat, lassen sich keine Antikörper gegen AChR oder MusK wie bei einem Großteil der Myasthenie-Patientinnen und Patienten nachweisen. Zudem sprechen die Betroffenen nicht auf eine immunsuppressive Therapie an.
Verlauf und Schweregrad der Erkrankung sind sehr unterschiedlich. Die Symptome können von kompletter Bewegungslosigkeit bei der Geburt bis hin zu nur leichter muskulärer Schwäche wie hängenden Augenlidern im Erwachsenenalter reichen. Eine gestörte Erregungsübertragung an der neuromuskulären Endplatte, bedingt durch Defekte in den beteiligten Proteinen, ist die Ursache der verschiedenen CMS-Formen. Die Formen unterscheiden sich je nach betroffenem Gen im Vererbungsmuster und im klinischen Bild, es stehen jeweils unterschiedliche Behandlungsoptionen zur Verfügung.
Oft können Patientinnen und Patienten trotz der Muskelschwäche mit einer geeigneten meist medikamentösen Therapie ein weitgehend normales Leben führen und auch ihren Beruf weiterhin ausüben. Dennoch ist mit Einschränkungen der körperlichen Belastbarkeit durch die Myasthenia gravis zu rechnen. Die Grenze der Belastbarkeit von an Myasthenie erkrankten Personen ist individuell unterschiedlich.
In der frühen Krankheitsphase ist auch eine Thymektomie (operative Entfernung der Thymusdrüse) eine mögliche Therapie der Myasthenia gravis. Dieser Eingriff am Thymus sollte individuell mit Patientinnen und Patienten, behandelnden Neurologinnen und Neurologen und Thoraxchirurg*innen besprochen werden. Dabei ist auch über verschiedene Verfahren der Operation am Thymus (transthorakal und minimalinvasiv) zu informieren. Eine Thymektomie ist bei Vorliegen eines Thymoms oder optional bei Patientinnen und Patienten mit generalisierter AChR-positiver MG sinnvoll. Seronegative MG-Patientinnen und Patienten (ohne nachweisbare Autoantikörper) sowie MuSK-positive MG-Patientinnen und Patienten scheinen nicht von einer Thymektomie zu profitieren.
Die medikamentöse Therapie der Myasthenia gravis basiert je nach Subtyp und Schweregrad der Erkrankung auf folgenden Medikamenten / Behandlungsstrategien:
Um eine optimale therapeutische Wirkung im Rahmen einer längerfristigen Therapie zu erzielen, müssen oft mehrere Medikamente und deren Kombinationen getestet werden.
Im Fall einer geplanten oder eingetretenen Schwangerschaft unter immunsuppressiver und symptomatischer Therapie oder Notwendigkeit einer solchen Therapie in der Schwangerschaft und Stillzeit wird ein Vorgehen gemäß aktueller Empfehlungen des Pharmakovigilanz- und Beratungszentrums für Embryonaltoxikologie unter www.embryotox.de empfohlen.
Patientinnen und Patienten mit Schluckbeschwerden und Tablettenaversion können von einer Medikamentengabe in flüssiger Form (Suspension) profitieren. Auch Betroffene mit Magensonde oder Intensivpatient*innen können mit einer Suspension versorgt werden, beispielsweise auch ältere PatientInnen mit einem schweren Verlauf und/oder einer Schluckstörung.
Ein Großteil der Myasthenie-Patientinnen und Patienten erfährt im Verlauf der Erkrankung mindestens einmal eine Schluckstörung , wobei der Zeitpunkt nicht genau vorherzusagen ist. Wann ein/e Erkrankte/r im Krankheitsverlauf eine Schluckstörung entwickelt, lässt sich nicht prognostizieren. Die Anzahl der dauerhaften Schluckstörungen bei Myastheniker*innen liegt ungefähr im einstelligen Prozentbereich.
Was ist eine myasthene Krise? Eine myasthene Krise ist ein schwerer Schub der Erkrankung. Auslöser können eine Infektion, psychische Belastung, Medikamenteneinnahmefehler oder unzureichende Immunsuppression sein. Bei einer myasthenen Krise nehmen die Symptome der Muskelschwäche so stark zu, dass die Schluck- und Atemmuskeln ihre Funktionen nicht mehr ausreichend erfüllen können. Dieser Zustand kann lebensbedrohlich werden. Die Mortalität bei einer myasthenen Krise liegt bei 5 %, sodass beim Auftreten schnell intensivmedizinisch reagiert werden muss.
Mögliche Auslöser einer myasthenen Krise können Infektionen, psychische Belastung, Medikamenteneinnahmefehler, unzureichende Immunsuppression oder deren zu frühes Absetzen sein. Besonders gefährdet sind Patientinnen und Patienten mit instabilen bulbären und respiratorischen Symptomen und multimorbide Patientinnen und Patienten im höheren Lebensalter.
Bulbäre Symptome sind sämtliche Beschwerden, die sich auf das Kauen, Schlucken und Sprechen beziehen, während respiratorische Symptome die Atmung betreffen. Kommt es also plötzlich zur akuten Atemnot, oder Patientinnen und Patienten sind von einem Tag auf den anderen nicht mehr in der Lage, selbstständig Nahrung herunterzuschlucken, oder sie zu zerkauen, liegt eine neurologische Notfallsituation vor, die einer intensivmedizinischen Behandlung bedarf.
Diese Maßnahmen können helfen, eine myasthene Krise zu vermeiden:
Verhalten im Ernstfall: Sollte eine myasthene Krise auftreten, sollten Betroffene diese sofort in einem Myasthenie-Zentrum, einer speziellen Myasthenie–Ambulanz oder einer neurologischen Klinik behandeln lassen. Bei Auftreten respiratorischer Insuffizienz und Aspiration sollte eine rasche intensivmedizinische Therapie inklusive Plasmaaustausch erfolgen.
Die Myasthenia gravis kann in unterschiedliche, autoimmune Formen unterteilt werden, je nach Vorkommen verschiedener Autoantikörper (AK). Die Therapie der MG orientiert sich neben der Krankheitsaktivität zunehmend am Antikörper-Status. Am entscheidendsten ist laut der überarbeiteten Leitlinien die Unterscheidung zwischen Formen der Erkrankung, bei denen nAChR-Antikörper auftreten und solchen, die mit der Bildung von MuSK-Antikörpern einhergehen. Bei Letzteren sind einige Medikamente zur Behandlung der Myasthenia gravis, nämlich die sogenannten C5-Komplementinhibitoren (z .B. Eculizumab und Ravulizumab), wirkungslos.
| Häufigkeit | Antikörper | |
|---|---|---|
| nAChR-AK | Ca. 80 – 85 % | Antikörper greifen die Acetylcholin-Rezeptoren an. |
| Anti-MuSK-AK (MAMG) | Ca. 5 % | IgG4-Antikörper stören die Interaktion zwischen LRP4 und der Tyrosinkinase, wodurch die Bildung der ACh-Rezeptoren gestört wird. |
| Anti-LRP4-AK | Ca. 2 % | Antikörper greifen das Transmembranprotein LRP4 (low-density lipoprotein receptor-like protein 4) an. |
| Anti-Titin-, Anti-Agrin- und Anti-RyR-AK | Ca. 3 % | Antikörper greifen verschiedene Enzyme und Rezeptoren an. Titin ist ein Strukturprotein der Muskeln. Agrin unterstützt die Bindung von ACh an die Acetylcholin-Rezeptoren. Ryanodin-Rezeptoren (RyR) befinden sich ebenfalls in den Muskeln und sind wichtig für die neuromuskuläre Übertragung. |
Die Early-onset MG trifft meist Frauen unter 45 Jahren.
• bei 20 % der MG-PatientInnen, Frauen 3x häufiger betroffen
• generalisierte Myasthenie mit frühem Erkrankungsbeginn unter 45 Jahren (Peak zw. 15-30 Jahren), (10 % sind unter 16 Jahren)
• Meist mit Thymus-Hyperplasie
• Meist mit Anti-AChR-AK
Late-onset MG betrifft größtenteils Männer ab 45 Jahren.
• Bei 45 %, Männer 5x häufiger betroffen
• generalisierte Myasthenie im höheren Lebensalter über 45 Jahren (Peak zw. 60 – 75 Jahren)
• Anti-AChR-AK und andere AK
Die Thymom-assoziierte MG kann jedes Lebensalter treffen, meist sind jedoch Männer wie Frauen zwischen 40 und 60 Jahren betroffen.
• 10-15 % haben ein Thymom (Tumor der Thymusdrüse)
• Anti-AChR-AK und andere AK, z.B. Titin-AK häufig bei Patient:innen unter 60 Jahren, Anti-MuSK-AK dagegen äußerst selten
Die Anti-MuSK-AK-assoziierte MG, bei der sich die Autoantikörper gegen die Muskel-spezifische Rezeptor-Tyrosinkinase (MuSK) richten, trifft meist Frauen jeden Lebensalters, jedoch oft eher jüngere Patientinnen.
• Bei 6 %, Frauen 3x häufiger betroffen
• Vor allem faziopharyngeale (Gesichts- und Rachen-)Muskeln betroffen
An der rein Okulären MG erkranken meist Frauen jeden Alters. Bei dieser Form sind insbesondere die äußeren Augenmuskeln betroffen, es treten Doppelbilder und herabhängende Oberlider auf.
• Bei 15 %
• 50-70 % mit Anti-AChR-AK
Zudem kann die Myasthenia gravis bei Neugeborenen auftreten. Die Neonatale Myasthenie kann durch Autoantikörper der Mutter bei Geburt oder beim Stillen übertragen werden.
Wie kommt es dazu?
Die Myasthenia gravis wirkt sich als chronische Erkrankung auf das alltägliche Leben der Betroffenen aus. Auch wenn mit der entsprechenden medikamentösen Therapie ein normaler Alltag in vielen Fällen weitestgehend möglich ist, können folgende Tipps den Patientinnen und Patienten helfen:
Das Lambert-Eaton-Myasthenie-Syndrom (LEMS) ist ebenfalls eine seltene Autoimmunerkrankung, die zu Myasthenie führt. Die Symptome der Myasthenia gravis und des LEMS sind sich also sehr ähnlich. Jedoch sind die Augen und die Atemmuskulatur sehr viel seltener betroffen. Stattdessen treten die Beschwerden vor allem an der rumpfnahen Bein- und Armmuskulatur auf.
Beide Erkrankungen beeinträchtigen also die Kommunikation zwischen Nerven und Muskeln. Doch die Ursachen sind andere. Die Antikörper, die beim LEMS gebildet werden, richten sich gegen die präsynaptischen Kalzium-Kanäle, ganz spezielle Strukturen an den Nervenendigungen. Dadurch wird die ACh-Ausschüttung generell reduziert, während bei der Myasthenia gravis die ACh-Rezeptoren geschädigt werden, wodurch der Botenstoff nicht mehr andocken kann. Auch wird ACh nicht schneller abgebaut, wie es bei Myasthenia gravis der Fall sein kann. Deshalb sind Cholinesterase-Hemmstoffe, anders als bei Myasthenia gravis, auch weniger wirksam bei der Behandlung der Symptome. Stattdessen wird primär 3,4-Diaminopyridin zur Behandlung eingesetzt.
S2k-Leitlinie Myasthenia gravis und Lambert-Eaton-Syndrom - Diagnostik und Therapie unter https://register.awmf.org/assets/guidelines/030-087l_S2k_Myasthenia_gravis_Lambert-Eaton-Syndrom_2017-03-abgelaufen.pdf - zuletzt abgerufen am 18.01.2023